As filed with the Securities and Exchange Commission on April 5, 2021
Registration No. 333-220368
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
POST-EFFECTIVE AMENDMENT NO. 3
TO
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
ADIAL PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | | 8071 | | 82-3074668 |
(State or other jurisdiction of
incorporation or organization) | | (Primary Standard Industrial
Classification Code Number) | | (I.R.S. Employer
Identification No.) |
1180 Seminole Trail, Suite 495
Charlottesville, Virginia 22901
(434) 422-9800
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive office)
William B. Stilley, III
President and Chief Executive Officer
Adial Pharmaceuticals, Inc.
1180 Seminole Trail, Suite 495
Charlottesville, Virginia 22901
(434) 422-9800
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Leslie Marlow, Esq.
Hank Gracin, Esq.
Patrick J. Egan, Esq.
Gracin & Marlow, LLP
The Chrysler Building
405 Lexington Avenue, 26th Floor
New York, NY 10174
Telephone: (212) 907-6457
Facsimile: (212) 208-4657
Approximate date of commencement of proposed sale to the public: From time to time after the effective date of this Registration Statement.
If any of the Securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, as amended, check the following box: ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act Registration Statement number of the earlier effective Registration Statement for the same offering: ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, please check the following box and list the Securities Act Registration Statement number of the earlier effective Registration Statement for the same offering: ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act Registration Statement number of the earlier effective Registration Statement for the same offering: ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ |
| Non-accelerated filer ☒ | Smaller reporting company ☒ |
| | Emerging Growth Company ☒ |
If an emerging growth company, indicate by checkmark if the registrant has not elected to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
EXPLANATORY NOTE
This Post-Effective Amendment No. 3 (this “Amendment”) to the Registration Statement on Form S-1, as amended by Amendment No. 1 and Amendment No. 2 thereto (Commission File No. 333-220368) (the “Original Registration Statement”), of Adial Pharmaceuticals, Inc. (the “Company”) is being filed pursuant to the undertakings in the Original Registration Statement to update and supplement the information contained in the Original Registration Statement, which was originally declared effective by the Securities and Exchange Commission on July 26, 2018.
The Original Registration Statement, as amended by this Amendment, pertains solely to the registration of an aggregate of 1,575,112 shares of common stock, par value $0.001 per share, consisting of: 1,516,552 shares of common stock underlying warrants (the “Investor Warrants”) previously issued by the Company to investors in its initial public offering and warrants to purchase 58,560 shares of common stock previously issued to the underwriters (the “Underwriters Warrants” and together with the Investor Warrants, the “Warrant”). The shares of Common Stock issuable upon exercise of the Warrants (the “Warrant Shares”) were initially registered on the Original Registration Statement.
For the convenience of the reader, this Amendment sets forth the Original Registration Statement in its entirety, as amended by this Amendment. This Amendment is being filed to incorporate certain information from the Company’s Annual Report on Form 10-K for the year ended December 31, 2020 filed with the SEC on March 22, 2021.
No additional securities are being registered under this Amendment. All applicable registration fees were paid at the time of the filing of the Original Registration Statement. Accordingly, the Company hereby amends the Original Registration Statement, as amended and supplemented through the date hereof, by filing this Amendment.
The information in this prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state or other jurisdiction where the offer or sale is not permitted.
| PROSPECTUS | SUBJECT TO COMPLETION, DATED APRIL 5, 2021 |

1,575,112 Shares of Common Stock Underlying Previously Issued Warrants
This prospectus relates to the offer and sale by Adial Pharmaceuticals, Inc., a Delaware corporation of up to 1,575,112 shares of common stock underlying warrants previously issued by us that are issuable at a price of $6.25 per share from time to time upon exercise of outstanding warrants issued to investors and the underwriters in our initial public offering, the issuance of which were previously registered on a Registration Statement on Form S-1 (File No. 333-220368).
We are not selling any shares of our common stock in this offering and, as a result, we will not receive any proceeds from the sale of the common stock covered by this prospectus. All of the net proceeds from the sale of our common stock will go to the warrant holders. Upon exercise of the warrants, however, we will receive proceeds from the exercise of such warrants if exercised for cash and not on a cashless basis. Any proceeds received from the exercise of such warrants will be used for general working capital and other corporate purposes.
Our common stock and the warrants issued in the initial public offering are listed on the Nasdaq Capital Market under the symbols “ADIL” and “ADILW.” On April 5, 2021, the last reported sale price of our common stock on the Nasdaq Capital Market was $2.44 per share. The last reported sale price of our warrants issued in the initial public offering on April 5, 2021 was $0.6435 per warrant. We urge prospective purchasers of our common stock to obtain current information about the market prices of our common stock and our warrants issued in connection with our initial public offering in July 2018.
We are an “emerging growth company” as that term is used in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), and, as such, elect to comply with certain reduced public company reporting requirements for future filings.
Investing in our securities involves significant risks, including those set forth in the “Risk Factors” section of this prospectus beginning on page 10.
See the “Plan of Distribution” section of this prospectus beginning on page 119 for more information on this offering.
No underwriter or person has been engaged to facilitate the sale of Shares in this offering. All costs associated with the registration were borne by us.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is April __, 2021
Table of Contents
You should rely only on the information contained in this prospectus and any free writing prospectus that we have authorized for use in connection with this offering. We have not authorized anyone to provide you with information that is different. We are offering to sell, and seeking offers to buy, the securities covered hereby only in jurisdictions where offers and sales are permitted. The information in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or any sale of the securities covered hereby. Our business, financial condition, results of operations and prospects may have changed since that date. We are not making an offer of these securities in any jurisdiction where the offer is not permitted. You should also read and consider the information in the documents to which we have referred you under the caption “Where You Can Find Additional Information” in the prospectus. In addition, this prospectus contains summaries of certain provisions contained in some of the documents described herein, but reference is made to the actual documents for complete information. All of the summaries are qualified in their entirety by the actual documents. Copies of some of the documents referred to herein have been filed, will be filed or will be incorporated by reference as exhibits to the registration statement of which this prospectus is a part, and you may obtain copies of those documents as described below under the heading “Where You Can Find Additional Information.”
For investors outside the United States: We have not done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside must inform themselves about, and observe any restrictions relating to, the offering of securities and the distribution of this prospectus outside the United States.
This prospectus includes statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. We believe that the data obtained from these industry publications and third-party research, surveys and studies are reliable. The Company is ultimately responsible for all disclosure included in this prospectus.
PROSPECTUS SUMMARY
The items in the following summary are described in more detail elsewhere in this prospectus and in the documents incorporated by reference herein. This summary highlights selected information contained elsewhere in this prospectus. This summary is not intended to be complete and does not contain all of the information that you should consider before deciding to invest in our securities. You should read this entire prospectus carefully, especially the “Risk Factors” section of this prospectus and other documents or information included or incorporated by reference in this prospectus before making an investment decision.
Overview
We are a clinical-stage biopharmaceutical company focused on the development of therapeutics for the treatment or prevention of addiction and related disorders. Our lead investigational new drug product, AD04, is being developed as a therapeutic agent for the treatment of alcohol use disorder (“AUD”). In January 2021, we expanded our portfolio in the field of addiction with the acquisition of Purnovate, LLC, and we continue to explore opportunities to expand our portfolio in the field of addiction and related disorders, both through internal development and through acquisitions. Our vision is to create the world’s leading addiction focused pharmaceutical company.
AUD is characterized by an urge to consume alcohol and an inability to control the levels of consumption. We have commenced the landmark ONWARD™ pivotal Phase 3 clinical trial using AD04 for the potential treatment of AUD in subjects with certain target genotypes. As of this filing, all 25 planned clinical sites were actively enrolling patients, and the ONWARD trial was more than 50% enrolled. The trial is expected to be completed by the first quarter of 2022. We believe our approach is unique in that it targets the serotonin system and individualizes the treatment of AUD, through the use of genetic screening (i.e., a companion diagnostic genetic biomarker). We have created an investigational companion diagnostic biomarker test for the genetic screening of patients with certain biomarkers that, as reported in the American Journal of Psychiatry (Johnson, et. al. 2011 & 2013), we believe will benefit from treatment with AD04. Our strategy is to integrate the pre-treatment genetic screening into AD04’s label to create a patient-specific treatment in one integrated therapeutic offering. Our goal is to develop a genetically targeted, effective and safe product candidate to treat AUD by reducing or eliminating the patients’ consumption of alcohol.
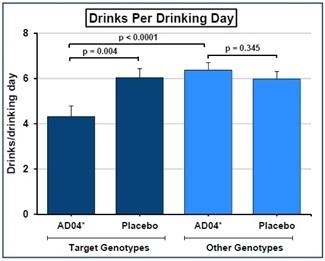
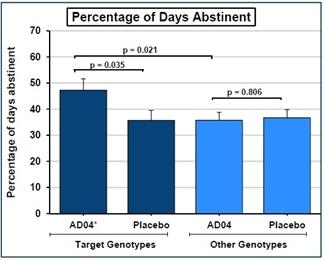
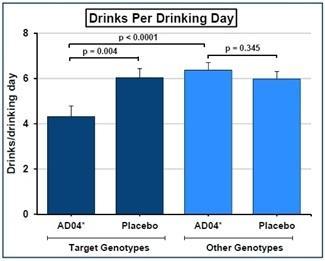
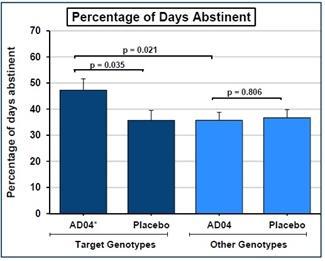
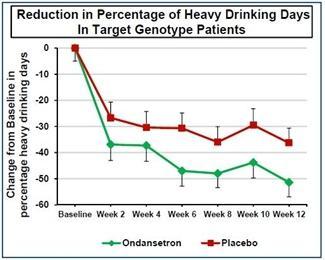
We have a worldwide, exclusive license from the University of Virginia Patent Foundation (d.b.a the Licensing & Venture Group) (“UVA LVG”), which is the licensing arm of the University of Virginia, to commercialize our investigational drug candidate, AD04, subject to Food and Drug Administration (“FDA”) approval of the product, based upon three separate patent application families, with patents issued in over 40 jurisdictions, including three issued patents in the U.S. Our investigational agent has been used in several investigator-sponsored trials and we possess or have rights to use toxicology, pharmacokinetic and other preclinical and clinical data that supports our landmark ONWARD pivotal Phase 3 clinical trial. Our therapeutic agent was the product candidate used in a University of Virginia investigator sponsored Phase 2b clinical trial of 283 patients. In this Phase 2b clinical trial, ultra-low dose ondansetron, the active pharmaceutical agent in AD04, showed a statistically significant difference between ondansetron and placebo for both the primary endpoint and secondary endpoint, which were reduction in severity of drinking measured in drinks per drinking day (1.71 drinks/drinking day; p=0.0042), and reduction in frequency of drinking measured in days of abstinence/no drinking (11.56%; p=0.0352), respectively. Additionally, and importantly, the Phase 2b results showed a significant decrease in the percentage of heavy drinking days (11.08%; p=0.0445) with a “heavy drinking day” defined as a day with four (4) or more alcoholic drinks for women or five (5) or more alcoholic drinks for men consumed in the same day.
The active pharmaceutical agent in AD04, our lead investigational new drug product, is ondansetron, which is also the active ingredient in Zofran®, which was granted FDA approval in 1991 for nausea and vomiting post-operatively and after chemotherapy or radiation treatment and is now commercially available in generic form. In studies of Zofran®, conducted as part of its FDA review process, ondansetron was given acutely at dosages up to almost 100 times the dosage expected to be formulated in AD04 with the highest doses of Zofran® given intravenously (“i.v.”), which results in approximately 160% of the exposure level as oral dosing. Even at high doses given i.v. the studies found that ondansetron is well-tolerated and results in few adverse side effects at the currently marketed doses, which reach more than 80 times the AD04 dose and are given i.v. The formulation dosage of ondansetron used in our drug candidate (and expected to be used by us in our Phase 3 clinical trials) has the potential advantage that it contains a much lower concentration of ondansetron than the generic formulation/dosage that has been used in prior clinical trials, is dosed orally, and is available with use of a companion diagnostic genetic biomarker. Our development plan for AD04 is designed to demonstrate both the efficacy of AD04 in the genetically targeted population and the safety of ondansetron when administered chronically at the AD04 dosage. However, to the best of our knowledge, no comprehensive clinical study has been performed to date that has evaluated the safety profile of ondansetron at any dosage for long-term use as anticipated in our ongoing and planned clinical trials.
AUD is characterized by an urge to consume alcohol and an inability to control the levels of consumption. Until the publication of the fifth revision of the Diagnostic and Statistical Manual of Mental Disorders in 2013 (the “DSM-5”), AUD was broken into “alcohol dependence” and “alcohol abuse”. More broadly, overdrinking due to the inability to moderate drinking is called alcohol addiction and is often called “alcoholism”, sometimes pejoratively.
Since ondansetron is already manufactured for generic sale, the active ingredient for AD04 is readily available from several manufacturers, and we have contracted with a U.S. manufacturer to acquire ondansetron at a cost expected to be under $0.01 per dose. Clinical trial material (“CTM”) has already been manufactured for the ONWARD Phase 3 trial. The CTM has demonstrated good stability after four years with the stability studies to date.
We have also developed the manufacturing process at a third-party vendor to produce tablets at what we expect will serve for commercial scale production (i.e., greater than 1 million tablets per batch), also at a cost expected to be less than $0.01 per dose. A proprietary packaging process has been developed, which appears to extend the stability of the drug product. Packaging costs are expected to be less than $0.05 per dose. We do not have a written commitment for supply of either the tablets or the packaging and believe that alternative suppliers are available to whom we can transfer the processes that have been developed.
Methods for the companion diagnostic genetic test have been developed as a blood test, and we established the test with a third-party vendor capable of supporting the ONWARD Phase 3 clinical trial. Additionally, we have built validation and possible approval of the companion diagnostic into the Phase 3 program, including that we plan to store blood samples for all patients in the event additional genetic testing is required by regulatory authorities.
Recent Developments
Clinical Developments
In January 2020, we announced that we had received favorable opinions from the Finnish Medicines Agency (FIMEA) and National Committee on Medical Research Ethics (TUKIJA) to commence our landmark ONWARD pivotal Phase 3 clinical trial to investigate AD04 as a genetically targeted therapeutic agent for the treatment of AUD.
On June 11, 2020, we announced that we had received all necessary approvals to commence our landmark ONWARD pivotal Phase 3 clinical trial to investigate AD04 as a genetically targeted therapeutic agent for the treatment of AUD.
On July 17, 2020, we received notice that the European Medicines Agency (EMA) had accepted the Pediatric Investigation Plan (PIP) submitted by us for development of our lead drug candidate, AD04, for the treatment of alcohol use disorder in the pediatric population, ages 12 to 17.
On July 30, 2020, we announced that we had received approval to commence our landmark ONWARD pivotal Phase 3 clinical trial of AD04 for the treatment of AUD in Croatia. As a result, we secured approvals to run the trial in all of the Scandinavian and Eastern European countries in which we intend to run the clinical trial.
On September 14, 2020, we filed with the FDA to take our Investigational New Drug (IND) Application for the use of our lead product candidate, AD04, as a treatment for Alcohol Use Disorder (AUD) off inactive status with the United States Food and Drug Administration (FDA) and on October 15, 2020 we announced that that the FDA had reactivated the IND.
On December 31, 2020, we provided the following highlighted updates on our landmark ONWARD pivotal Phase 3 clinical trial of AD04 for the treatment of AUD.
| | ● | All 25 planned investigative sites are active. |
| | ● | 35% of the planned 290 subjects in the ONWARD trial were enrolled |
| | ● | The enrollment rate has increased for four consecutive months and the early termination rate has been lower than expected. |
| | ● | AD04 has been well tolerated with no serious adverse events having been reported |
Acquisition of Purnovate, LLC
On January 26, 2021, we closed the acquisition (the “Acquisition”) contemplated by that certain Equity Purchase Agreement, dated December 7, 2020, as amended (the “Purchase Agreement”), by and among Adial, Purnovate, LLC (“Purnovate”), each of the members of Purnovate (the “Members”) and Dr. Robert D. Thompson, as representative of the Members.
Prior to closing, we had advanced Purnovate $350,000 for use as working capital during the due diligence period. At closing, in exchange for Purnovate, Adial paid the members an additional $350,000 (the “Cash Consideration”) and issued to the members an aggregate of 700,000 shares of Adial restricted common stock (the “Stock Consideration”) with an approximate total market value of $1,638,000. In addition, members will receive (i) development milestone payments in an aggregate amount of up to $2,100,000 for each compound developed, (ii) development milestone payments in an aggregate amount of up to $20,000,000 for each compound commercialized, and (iii) royalties of 3.0% of Net Sales (as such term is defined in the Purchase Agreement). The Stock Consideration has been placed into escrow to secure certain indemnification and other obligations of Purnovate and the members in connection with the Acquisition.
The acquisition was effected by a merger (the “Merger”) of Purnovate into Purnovate, Inc., a Delaware corporation and wholly owned subsidiary of Adial, which survived the Merger. In connection with the Merger, on January 20, 2021, Purnovate converted from a limited liability company to a corporation and on January 25, 2021, the parties entered into an Amendment to the Purchase Agreement (the “Amendment”) to provide for the mechanism of closing the Acquisition through a Merger.
Purnovate is a drug development company with a platform focused on developing drug candidates for non-opioid pain reduction and other diseases and disorders potentially targeted with adenosine analogs that are selective, potent, stable, and soluble. Purines are a class of chemical structures that include adenosine, an important neurotransmitter. Purnovate uses innovative methods and technologies to enhance the drug properties of purines, which are a class of chemical structures that include adenosine, an important neurotransmitter. Adenosine receptors are hypothesized to be involved in the mediation of nociception (i.e., pain) and blocking the receptor is proposed as an anti-nociceptive. Additionally, selective adenosine analogs may be useful to treat pain while avoiding the tolerability problems (i.e., wakefulness, gastrointestinal) and safety issues (e.g., cardiac block and vasodilation) and therefore have the potential to provide a meaningful non-opioid treatment for pain without the side effects of earlier generations of adenosine compounds or certain other pain products. These adenosine analogs may also be useful in a number of other diseases and disorders such as cocaine addiction, infectious disease, inflammation, cancer, asthma, and diabetes. Purnovate’s portfolio of pre-clinical technologies are also believed to offer opportunities to improve the characteristics of other classes of molecules outside of the adenosine chemistry space and maybe even outside the purine chemistry space. All drug candidates developed using Purnovate’s platform technologies are expected to be patently distinct new chemical entities (i.e., patentable compositions of matter). Purnovate operates a chemistry and analytics laboratory in its 4,175 square feet leased laboratory and office space. Purnovate has been synthesizing new adenosine analog chemical entities with promising potency, selectivity, stability, and solubility characteristics.
Our Strategy
We develop pharmaceutical treatments for addictions and addictive disorders and related diseases and disorders. Our business strategy is to advance AD04, our lead investigational drug candidate, toward regulatory approval for alcohol addiction in the United States, the European Union, and then eventually other territories. We subsequently plan to develop label expansions into other indications (e.g., opioid use disorder, other drug addictions, obesity, smoking cessation, eating disorders and anxiety). Additionally, we are inventing and developing novel therapeutic agents at our chemistry facilities and seeking to acquire addiction related assets, particularly those expected to be synergistic with AD04 once it is marketed, if it is approved.
Our goals in executing this strategy are to keep capital requirements to a minimum, expedite product development, gain access to clinical research and manufacturing expertise that will advance product development, approval and eventual market uptake of our product, and rely on a well-defined and carefully executed intellectual property strategy in order to position our products with long-term, defensible, competitive advantages. Execution of this strategy may include seeking grant funding and funding from partners and collaborators when available on terms we believe to be favorable to us, and on which there is no guarantee will be available. In collaboration with our CRO, we have been and are working to adapt the implementation of our strategy in response to the ongoing coronavirus pandemic.
Our near-term strategy includes:
| | ● | Obtaining regulatory approval for our lead product in the United States and Europe. We have commenced our initial Phase 3 clinical trial for the treatment of AUD in Scandinavia and Central and Eastern Europe. If our initial Phase 3 clinical trial is successful, we expect to conduct a second, and possibly a third, Phase 3 clinical trial in the same areas but with additional clinical sites in the United States and Western Europe. |
| | ● | Prosecuting and expanding our intellectual property and product portfolio. We have acquired rights to a promising drug candidate and made a significant investment in the development of our licensed patent portfolio to protect our technologies and programs, and we intend to continue to do so. We have obtained exclusive rights to three different patent families directed to therapeutic methods related to our AD04 platform. These families include 3 issued U.S. patents, and at least one foreign equivalent patent covering AD04 issued in over 40 national jurisdictions, including most of Europe and Eurasia. Divisional and continuation applications to expand the coverage have also been filed in certain jurisdictions. Additionally, commencing in early 2021, we have an adenosine platform that has and is expected to continue to generate what we believe are patentable new chemical entities. We intend that further product portfolio expansions will be focused on promising addiction therapies and/or late-stage clinical assets. |
| | | |
| | ● | Evaluating the additional use of our product candidate in other indications. In addition to alcohol addiction, we plan to conduct exploratory work to investigate using AD04 as a potential treatment for opioid use disorder, gambling addiction, smoking cessation, obesity, and other addiction related disorders in which 5-HT3 antagonism may have a treatment effect. We believe we will be able to undertake this initial exploratory effort with minimal additional cash cost to our company through the use of academic partnerships, grants, human laboratory studies and/or non-clinical studies. We believe that, due to its hypothesized mechanism of action (i.e., the modulation of the serotonin system in patients that are genetically targeted based on the apparent sensitivity to such modulation, where the modulation appears to reduce cravings), AD04 has the potential to be used for the treatment of such other addictive disorders. To date, we have not discussed these potential uses with the FDA or any other regulatory bodies. |
| | ● | Maximizing commercial opportunity for our technology. AD04 targets large markets with significant unmet medical need. We intend to develop an extended release, once-a-day formulation of AD04 to enhance compliance and market appeal. |
| | | |
| | ● | Managing our business with efficiency and discipline. We believe we have efficiently utilized our capital and human resources to develop and acquire our product candidate and programs and create a broad intellectual property portfolio. We operate cross-functionally and are led by an experienced management team with backgrounds in developing product candidates. We use project management techniques to assist us in making disciplined strategic program decisions and to attempt to limit the risk profile of our product pipeline. |
Summary Risk Factors
Our business faces significant risks and uncertainties of which investors should be aware before making a decision to invest in our common stock. If any of the following risks are realized, our business, financial condition and results of operations could be materially and adversely affected. The following is a summary of the more significant risks relating to our Company. A more detailed description of our risk factors set forth under the caption “Risk Factors” beginning on page 10.
Risks Relating to our Company
| | ● | We have a limited operating history with which to compare, have incurred significant losses since our inception, and expect to incur substantial and increasing losses for the foreseeable future. |
| | ● | We currently have no product revenues and may not generate revenue at any time in the near future, if at all. |
| | ● | We and our independent registered accounting firm has expressed substantial doubt about our ability to continue as a going concern. |
| | ● | We will need to secure additional financing, which may not be available to us on favorable terms, if at all. |
| | ● | We have identified weaknesses in our internal controls, and it cannot be assured that these weaknesses will be effectively remediated or that additional material weaknesses will not occur in the future. |
| | ● | We rely on a license to use various technologies that are material to our business and there is no guarantee that such license agreements won’t be terminated, or that other rights necessary to commercialize our products will be available to us on acceptable terms or at all. |
| | ● | Our business is dependent upon the success of our lead product candidate, AD04, which requires significant additional clinical testing before we can seek regulatory approval and potentially launch commercial sales. |
| | ● | The active ingredient of our product candidate, ondansetron, is currently available in generic form and has been shown to have adverse effects on patients. |
| | ● | Coronavirus or other global health crises could adversely impact our business, including our clinical trials. |
| | ● | While there exists a large body of evidence supporting the safety of our primary API, ondansetron, under short-term use, there are currently no long-term use clinical safety data available. |
| | ● | All of our current data for our lead product candidate are the result of Phase 2 clinical trials conducted by third parties and do not necessarily provide sufficient evidence that our products are viable as potential pharmaceutical products. |
| | ● | The FDA and/or EMA may not accept our planned Phase 3 endpoints for final approval of AD04 and may determine additional clinical trials are required for approval of AD04. |
| | ● | Our lead investigational product, AD04, is dependent on a successful development, approval, and commercialization of a genetic test, which is expected to be classified as a companion diagnostic, which may not attain regulatory approval. |
| | ● | We have limited experience as a company conducting clinical trials, any may experience delays in our clinical trials and may fail to demonstrate adequately the safety and efficacy of AD04 or any future product candidates. |
| | ● | Our product candidate and the product candidates of Purnovate are in the early stages of development and there is uncertainty as to market acceptance of our technology and product candidates. |
Risks Relating to our Acquisition of Purnovate
| | ● | The combined company may not experience the anticipated strategic benefits of the Acquisition and we may be unable to successfully integrate the Purnovate businesses. |
| | ● | Purnovate has a limited operating history upon which to evaluate its ability to commercialize its products. |
Risks Relating to our Business and Industry
| | ● | We must obtain and maintain regulatory approvals in every jurisdiction in which we intend to sell our product candidate and the regulatory approval in one jurisdiction does not guarantee the approval in another jurisdiction. |
| | ● | AD04 and any future product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential or result in significant negative consequences such as incurring product liability lawsuits |
| | ● | We will continue to be subject to ongoing and extensive regulatory requirements even after regulatory approval, and compliance with such regulatory requirements cannot be assured. |
| | ● | Our employees, independent contractors, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements. |
| | ● | We have no experience selling, marketing or distributing products and have no internal capability to do so. |
| | ● | We may not be successful in establishing and maintaining strategic partnerships. |
| | ● | We have limited protection for our intellectual property. Our licensed patents and proprietary rights may not prevent us from infringing on the rights of others or prohibit potential competitors from commercializing products. |
| | ● | We may be involved in lawsuits to protect or enforce the patents of our licensors, which could be expensive, time-consuming and unsuccessful. |
| | ● | We rely on key executive officers and scientific, regulatory and medical advisors, and their knowledge of our business and technical expertise would be difficult to replace. |
| | ● | Certain of our officers may have a conflict of interest. |
Risks Related to our Securities and Investing in our Securities
| | ● | Certain of our shareholders have sufficient voting power to make corporate governance decisions that could have a significant influence on us and the other stockholders. |
| | ● | We have never paid dividends and have no plans to pay dividends in the foreseeable future. |
| | ● | Our failure to meet the continued listing requirements of The Nasdaq Capital Market could result in a de-listing of our common stock. |
| | ● | Our stock price has fluctuated in the past, has recently been volatile and may be volatile in the future, and as a result, investors in our common stock could incur substantial losses. |
| | ● | Our need for future financing may result in the issuance of additional securities which will cause investors to experience dilution. |
| | ● | Provisions in our corporate charter documents and under Delaware law could make an acquisition of our company, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management. |
| | ● | Our Certificate of Incorporation and our bylaws provide that the Court of Chancery of the State of Delaware will be the exclusive forum for certain types of state actions that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, or employees. |
| | ● | If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline. |
Implications of Being an Emerging Growth Company
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), and therefore we intend to take advantage of certain exemptions from various public company reporting requirements, including not being required to have our internal controls over financial reporting audited by our independent registered public accounting firm pursuant to Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and any golden parachute payments. We may take advantage of these exemptions until we are no longer an “emerging growth company.” In addition, the JOBS Act provides that an “emerging growth company” can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have elected to use the extended transition period for complying with new or revised accounting standards under the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates. We will remain an “emerging growth company” until the earlier of (1) the last day of the fiscal year: (a) following the fifth anniversary of the completion of our initial public offering; (b) in which we have total annual gross revenue of at least $1.07 billion; or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeded $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period. References herein to “emerging growth company” have the meaning associated with that term in the JOBS Act.
Corporate Information
ADial Pharmaceuticals, L.L.C. was formed as a Virginia limited liability company in November 2010. ADial Pharmaceuticals, L.L.C. converted from a Virginia limited liability company into a Virginia corporation on October 3, 2017, and then reincorporated in Delaware on October 11, 2017 by merging the Virginia corporation with and into Adial Pharmaceuticals, Inc., a Delaware corporation that was incorporated on October 5, 2017 as a wholly owned subsidiary of the Virginia corporation. We refer to this as the corporate conversion/reincorporation. In connection with the corporate conversion/reincorporation, each unit of ADial Pharmaceuticals, L.L.C. was converted into shares of common stock of the Virginia corporation and then into shares of common stock of Adial Pharmaceuticals, Inc., the members of ADial Pharmaceuticals, L.L.C. became stockholders of Adial Pharmaceuticals, Inc. and Adial Pharmaceuticals, Inc. succeeded to the business of ADial Pharmaceuticals, L.L.C.
Our principal executive offices are located at 1180 Seminole Trail, Suite 495, Charlottesville VA 22901, and our telephone number is (434) 422-9800. Our website address is www.adialpharma.com. Information contained in our website does not form part of prospectus and is intended for informational purposes only.
This prospectus contains references to our trademarks and to trademarks belonging to other entities. Solely for convenience, trademarks and trade names referred to in this prospectus, including logos, artwork and other visual displays, may appear without the ® or TM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
THE OFFERING
Common Stock Offered by us | | Up to 1,575,112 shares of common stock issuable upon exercise of the outstanding warrants issued in our IPO. Each warrant is exercisable at any time for the purchase of one share of our common stock at an exercise price of $6.25 per share. The warrants expire on July 31, 2023. |
| | | |
Common Stock Outstanding prior to this offering Common Stock Outstanding after this offering | | 17,269,877(1) 18,844,989 (assuming full exercise of the warrants issued in our initial public offering) |
| | | |
| Use of Proceeds | | We will receive approximately $9,844,450 of proceeds if all the currently outstanding warrants issued in the IPO are exercised for cash. We currently intend to use these proceeds for general corporate purposes. See “Use of Proceeds.” |
| | | |
| NASDAQ Capital Markets Symbols | | Our common stock and the warrants issued in the IPO are listed on the Nasdaq Capital Market under the symbols “ADIL” and “ADILW,” respectively. |
| | | |
Risk Factors | | Investment in our securities involves a high degree of risk and could result in a loss of your entire investment. See “Risk Factors” beginning on page 10 to read about factors you should consider before buying our shares of common stock. |
| (1) | The number of shares of our common stock that will be outstanding immediately after this offering is based on 17,269,877 shares of common stock outstanding as of March 22, 2021. |
Unless specifically stated otherwise, the information in this prospectus:
| | ● | excludes an additional 7,957,225 shares of our common stock issuable upon the exercise of outstanding warrants at a weighted average exercise price of $4.83 per share; |
| | ● | excludes an additional 3,576,866 shares of our common stock issuable upon outstanding options to purchase shares of common stock with a weighted average exercise price of $2.61 per share; and |
| | | |
| | ● | excludes an additional 1,028,382 shares of our common stock reserved for future issuance under the 2017 equity incentive plan. |
OUR INITIAL PUBLIC OFFERING
On July 31, 2018, we closed our IPO whereby we sold 1,464,000 units, each unit consisting of one share of common stock and one warrant to purchase one share of common stock, at a public offering price of $5.00 per unit, before underwriting discounts and expenses. In addition, the underwriters partially exercised their over-allotment option to purchase up to 219,600 warrants granted in connection with the offering by purchasing an additional 170,652 warrants at the offering price of $0.01 per warrant, for proceeds of $1,707. The aggregate net proceeds received by us from the offering were $6.3 million, net of offering expenses not recognized in previous periods. Approximately $633,000 of these proceeds were used for cash repayment of debts. We also issued to the underwriters in the IPO warrants to purchase 58,560 shares of common stock at a purchase price of $6.25 per share.
As of the date of this prospectus, we had outstanding warrants to purchase an aggregate of 1,575,112 shares of our common stock, which warrants were issued in the IPO, calculated as follows: (i) 1,516,552 shares of our common stock issuable upon exercise of warrants issued to investors and (ii) 58,560 shares of our common stock issuable upon exercise of warrants issued to the representative of the underwriters.
RISK FACTORS
Investors should carefully consider the risks described below before deciding whether to invest in our securities. If any of the following risks actually occurs, our business, financial condition or results of operations could be adversely affected. In such case, the trading price of our common stock could decline and you could lose all or part of your investment. Our actual results could differ materially from those anticipated in the forward-looking statements made throughout this prospectus as a result of different factors, including the risks we face described below.
Risks Relating to our Company
We have incurred net losses every year and quarter since our inception and anticipate that we will continue to incur net losses in the future.
We are a clinical stage biotechnology pharmaceutical company that is focused on the discovery and development of medications for the treatment of addictions and related disorders of AUD in patients with certain targeted genotypes. We have a limited operating history. Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate effect or an acceptable safety profile, gain regulatory approval and become commercially viable. We have no products approved for commercial sale and have not generated any revenue from product sales to date, and we continue to incur significant research and development and other expenses related to our ongoing operations. To date, we have not generated positive cash flow from operations, revenues, or profitable operations, nor do we expect to in the foreseeable future. As of December 31, 2020, we had an accumulated deficit of approximately $31.5 million.
Even if we succeed in commercializing our product candidate or any future product candidates, we expect that the commercialization of our product will not begin until 2024 or later, we will continue to incur substantial research and development and other expenditures to develop and market additional product candidates and will continue to incur substantial losses and negative operating cash flow. We may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenue. Our prior losses and expected future losses have had and will continue to have an adverse effect on our shareholders’ equity and working capital.
We currently have no product revenues and may not generate revenue at any time in the near future, if at all. Currently, we have no products approved for commercial sale.
We currently have no products for sale and we cannot guarantee that we will ever have any drug products approved for sale. We and our product candidate are subject to extensive regulation by the FDA, and comparable regulatory authorities in other countries governing, among other things, research, testing, clinical trials, manufacturing, labeling, promotion, marketing, adverse event reporting and recordkeeping of our product candidates. Until, and unless, we receive approval from the FDA or other regulatory authorities for our product candidates, we cannot commercialize product candidates and will not have product revenues. Even if we successfully develop products, achieve regulatory approval, and then commercialize our products, we may be unable to generate revenue for many years, if at all. We do not anticipate that we will generate revenue for at least several years, if at all. If we are unable to generate revenue, we will not become profitable, and we may be unable to continue our operations. For the foreseeable future, we will have to fund all of our operations from equity and debt offerings, cash on hand and grants. In addition, changes may occur that would consume our available capital at a faster pace than expected, including changes in and progress of our development activities, acquisitions of additional candidates and changes in regulation. Moreover, preclinical and clinical testing may not start or be completed as we forecast and may not achieve the desired results. Therefore, we expect to seek additional sources of funding, such as additional financing, grant funding or partner or collaborator funding, which additional sources of funding may not be available on favorable terms, if at all.
We have had limited operations to date and there can be no assurance that we will be able to execute on our business strategy.
We are a clinical stage company and have had limited operations to date. We have yet to demonstrate our ability to overcome the risks frequently encountered in our industry and are still subject to many of the risks common to such enterprises, including our ability to implement our business plan, market acceptance of our proposed business and lead product, under-capitalization, cash shortages, limitations with respect to personnel, financing and other resources, competition from better funded and experienced companies, and uncertainty of our ability to generate revenues. In fact, though individual team members have experience running clinical trials, as a company we have yet to prove that we can successfully run a clinical trial. There is no assurance that our activities will be successful or will result in any revenues or profit, and the likelihood of our success must be considered in light of the stage of our development. In addition, no assurance can be given that we will be able to consummate our business strategy and plans, or that financial, technological, market, or other limitations may force us to modify, alter, significantly delay, or significantly impede the implementation of such plans. We have insufficient results for investors to use to identify historical trends. Investors should consider our prospects in light of the risk, expenses and difficulties we will encounter as an early stage company. Our revenue and income potential is unproven and our business model is continually evolving. We are subject to the risks inherent to the operation of a new business enterprise, and cannot assure you that we will be able to successfully address these risks.
We and our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern.
We have suffered recurring losses from operations based on our development plans and our operating requirements. These conditions, among others, considered in the aggregate raise substantial doubt about the Company’s ability to continue as a going concern for at least one year from the issuance of the accompanying financial statements. Although the funds raised as a result of the completion of our IPO, the receipt of proceeds from the exercise of warrants, and completion of various follow on financings have sustained our operations through 2020, based on our current development plans and operating requirements, we project that, without additional funding we will have fully expended our funds in the fourth quarter of 2021. We have executed an equity purchase agreement (the “Equity Purchase Agreement”) with Keystone Capital, LLC (“Keystone Capital”), which allows us to obtain, depending on our stock’s market price and fulfillment of certain conditions, up to $15 million in equity financing, of which we have already raised $2.0 million. However, since the market price of our stock is volatile, we cannot state with certainty how much, if any, of the remaining funding will be available. The actual number of shares that are sold to Keystone Capital may depend based on a number of factors, including the market price of the common stock during the sales period, during which sales cannot take place if the market price of a share of our common stock is below one dollar. Actual gross proceeds may be less than $15.0 million, which may impact our future liquidity. Because the price per share of each share sold to Keystone Capital will fluctuate during the sales period, it is not currently possible to predict the number of shares that will be sold or the actual gross proceeds to be raised in connection with those sales. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty. If we cannot raise the necessary capital to continue as a viable entity, we could experience a material adverse effect on our business and our stockholders may lose some or all of their investment in us.
We will need to secure additional financing in order to support our operations and fund our current and future clinical trials. We can provide no assurances that any additional sources of financing will be available to us on favorable terms, if at all. Our forecast of the period of time through which our current financial resources will be adequate to support our operations and the costs to support our general and administrative, selling and marketing and research and development activities are forward-looking statements and involve risks and uncertainties.
If we do not succeed in raising additional funds on acceptable terms, we may be unable to complete planned product development activities or obtain approval of our product candidate from the FDA and other regulatory authorities. We do not have any committed sources of capital other than our equity line with Keystone Capital for which there can be no assurance that we will meet the use requirements. Moreover, if our trial activities are significantly delayed due to the coronavirus pandemic, we would not be able to reach database lock with cash on hand even with receipt of the grants to which we have applied. In such case, we would need to obtain additional funding, either through other grants or through potentially dilutive means. In any case, we will need to raise additional capital to complete our development program and to meet our long-term business objectives.
Cash and cash equivalents at the date of this prospectus will not be sufficient to fund our operations for the next twelve months, given current expectations. We will require additional financing as we continue to execute our business strategy, including that we will require additional funds in order for additional Phase 3 trials of AD04, as well as any additional clinical trials or other development of any products we may acquire or license, including those acquired from Purnovate. Our liquidity may be negatively impacted as a result of a research and development cost increases in addition to general economic and industry factors. We anticipate that, to the extent that we require additional liquidity, it will be funded through the incurrence of other indebtedness, additional equity financings or a combination of these potential sources of liquidity. In addition, we may raise additional funds to finance future cash needs through grant funding and/or corporate collaboration and licensing arrangements. If we raise additional funds by issuing equity securities or convertible debt, including pursuant to our Equity Purchase Agreement with Keystone Capital, our stockholders will experience dilution. Debt financing, if available, would result in increased fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our products, future revenue streams or product candidates or to grant licenses on terms that may not be favorable to us. The covenants under future credit facilities may limit our ability to obtain additional debt financing. We cannot be certain that additional funding will be available on acceptable terms, or at all. Any failure to raise capital in the future could have a negative impact on our financial condition and our ability to pursue our business strategies.
Additional financing, which is not in place at this time, may be from the sale of equity or convertible or other debt securities in a public or private offering, from a credit facility or strategic partnership coupled with an investment in us or a combination of both. Our ability to raise capital through the sale of equity may be limited by the various rules of the Securities and Exchange Commission (the “SEC”) and The Nasdaq Capital Market (the “Nasdaq”), which place limits on the number of shares of stock that may be sold. Equity issuances would have a dilutive effect on our stockholders. We may be unable to raise sufficient additional financing on terms that are acceptable to us, if at all. Our failure to raise additional capital and in sufficient amounts may significantly impact our ability to expand our business. For further discussion of our liquidity requirements as they relate to our long-term plans, see the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources.”
We have identified weaknesses in our internal controls, and we cannot provide assurances that these weaknesses will be effectively remediated or that additional material weaknesses will not occur in the future.
As a public company, we are subject to the reporting requirements of the Exchange Act, and the Sarbanes-Oxley Act. We expect that the requirements of these rules and regulations will continue to increase our legal, accounting and financial compliance costs, make some activities more difficult, time consuming and costly, and place significant strain on our personnel, systems and resources.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures, and internal controls over financial reporting.
We do not yet have effective disclosure controls and procedures, or internal controls over all aspects of our financial reporting. We are continuing to develop and refine our internal controls over financial reporting. Our management is responsible for establishing and maintaining adequate internal control over our financial reporting, as defined in Rule 13a-15(f) under the Exchange Act. We will be required to expend time and resources to further improve our internal controls over financial reporting, including by expanding our staff. However, we cannot assure you that our internal control over financial reporting, as modified, will enable us to identify or avoid material weaknesses in the future.
We have identified material weaknesses in our internal control over financial reporting. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis. The material weaknesses identified to date include (i) policies and procedures which are not yet adequately documented, (ii) approval processes and review processes and documentation for such reviews, (iii) GAAP experience regarding complex transactions and reporting, and (iv) optimal segregation of duties and levels of oversight. As such, our internal controls over financial reporting were not designed or operating effectively.
We will be required to expend time and resources to further improve our internal controls over financial reporting, including by expanding our staff. However, we cannot assure you that our internal control over financial reporting, as modified, will enable us to identify or avoid material weaknesses in the future.
Our current controls and any new controls that we develop may become inadequate because of changes in conditions in our business, including increased complexity resulting from our international expansion. Further, weaknesses in our disclosure controls or our internal control over financial reporting may be discovered in the future. Any failure to develop or maintain effective controls, or any difficulties encountered in their implementation or improvement, could harm our operating results or cause us to fail to meet our reporting obligations and may result in a restatement of our financial statements for prior periods. Any failure to implement and maintain effective internal control over financial reporting could also adversely affect the results of management reports and independent registered public accounting firm audits of our internal control over financial reporting that we will eventually be required to include in our periodic reports that will be filed with the SEC. Ineffective disclosure controls and procedures, and internal control over financial reporting could also cause investors to lose confidence in our reported financial and other information, which would likely have a negative effect on the market price of our common stock.
Our independent registered public accounting firm is not required to audit the effectiveness of our internal control over financial reporting until after we are no longer an “emerging growth company” as defined in the JOBS Act and meet other requirements. At such time, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied with the level at which our internal control over financial reporting is documented, designed or operating. Any failure to maintain effective disclosure controls and internal control over financial reporting could have a material and adverse effect on our business and operating results, and cause a decline in the market price of our common stock.
We rely on a license to use various technologies that are material to our business and if the agreement were to be terminated or if other rights that may be necessary or we deem advisable for commercializing our intended products cannot be obtained, it would halt our ability to market our products and technology, as well as have an immediate material adverse effect on our business, operating results and financial condition.
Our prospects are significantly dependent upon the UVA LVG License. The UVA LVG License grants us exclusive, worldwide rights to certain existing patents and related intellectual property that covers AD04, our lead and currently only product candidate. If we breach the terms of the UVA LVG License, including any failure to make minimum royalty payments required thereunder or failure to reach certain developmental milestones and completion of deadlines, including, submitting an NDA by December 31, 2024 and commencing commercialization of an FDA approved product by December 31, 2025, or other factors, including but not limited to, the failure to comply with material terms of the Agreement, the licensor has the right to terminate the license. If we were to lose or otherwise be unable to maintain this license on acceptable terms, or find that it is necessary or appropriate to secure new licenses from other third parties, we would not be able to market our products and technology, which would likely require us to cease our current operations which would have an immediate material adverse effect on our business, operating results and financial condition.
Our business is dependent upon the success of our lead product candidate, AD04, which requires significant additional clinical testing before we can seek regulatory approval and potentially launch commercial sales. We do not have any other products in clinical development.
Our business and future success depends upon our ability to obtain regulatory approval of and then successfully commercialize our lead investigational product candidate, AD04. AD04 is in clinical stage development. To date, our main focus and the investment of a significant portion of our efforts and financial resources has been in the development of our lead and only investigational product candidate, AD04, for which we are currently conducting the ONWARD Phase 3 clinical trial with approximately 290 patients in Scandinavia and Central and Eastern Europe, which targets the reduction of risk drinking (heavy drinking of alcohol) in subjects that possess selected genetics of the serotonin transporter and/or 5-HT3 receptor gene. We expect that at least one additional Phase 3 clinical trial will be required for approval, as well as, one or more supportive clinical studies Even though we are pursuing a registration pathway based on specific FDA input and guidance and the EMA precedents and guidance, there are many uncertainties known and unknown that may affect the outcome of the trial. These include adequate patient enrollment, adequate supply of our product candidate, potential changes in the regulatory landscape, and the results of the trial being successful.
All of our future product candidates, as well as AD04, will require additional clinical and non-clinical development, regulatory review and approval in multiple jurisdictions, substantial investment, access to sufficient commercial manufacturing capacity and significant marketing efforts before we can generate any revenue from product sales. We expect AD04 will need at least two Phase 3 trials (including the ONWARD Phase 3 trial are conducting in Scandinavia and Central and Eastern Europe) and one or more supportive clinical studies to gain approval in either the U.S. or Europe for AUD and additional development activity, including, without limitation, clinical trials, in order to seek approval for the use of AD04 to treat any other indications (e.g., such as opioid use disorder, gambling addiction, smoking cessation, and other drug addictions). In addition, because AD04 is our most advanced product candidate and there is limited history information on long-term effects of our proposed dosage, there is always a chance of developmental delays or regulatory issues or other problems arising, with our development plans and depending on their magnitude, our business could be significantly harmed. In any case, the costs associated with completion of our ONWARD Phase 3 trial, a second, confirmatory trial, commercialization of AD04, and the costs of developing AD04 for use in other indications are significant, and will require obtaining funding, possibly through equity sales, before AD04 generates revenue.
We may apply to the FDA for the AD04 program to be designated as a fast track development program, accelerated approval, priority review, or breakthrough therapy designation. Designation as a fast track development program, accelerated approval, priority review, or breakthrough therapy designation is within the discretion of the FDA. Accordingly, even if we believe that one of our product candidates meets the criteria for designation as a fast track development program, accelerated approval, priority review, or breakthrough therapy designation, the FDA may disagree and instead determine not to make such designation. Even if we receive a fast track development program, accelerated approval, priority review, or breakthrough therapy designation, the receipt of such designation or approval for a product candidate may not result in a faster development of any product candidate or approval process for product candidate. In addition, even if one or more of our product candidates qualify as fast track development program, accelerated approval, priority review, or breakthrough therapy designation, the FDA may later decide that the product candidates no longer meet the conditions for qualification or decide that the time period for FDA review or approval will not be shortened and the designation may result in no benefit to the Company.
Our future success depends heavily on our ability to successfully manufacture, develop, obtain regulatory approval, and commercialize AD04, which may never occur. We currently generate no revenues from our product candidate, and we may never be able to develop or commercialize a marketable drug.
The active ingredient of our product candidate, ondansetron, is currently available in generic form.
Ondansetron, the active pharmaceutical ingredient (“API”) of our current drug treatment, was granted FDA approval as Zofran® in January 1991 and is approved in many foreign markets. Ondansetron is commercially available in generic form, but not available: (i) at the formulation/dosage levels expected to be marketed by us, or (ii) with a requirement to use a diagnostic biomarker, as we expect to be the case with AD04. Although ondansetron has been approved to treat nausea and emesis it has not been approved to treat AUD and it has not been approved for daily long-term use as planned by us. Clinical testing to date of ondansetron at the higher doses used to treat nausea/emesis have not shown effectiveness in treating AUD or any other addictive disorder; however, if a third party conducted a Phase 3 clinical program and showed success treating AUD at those doses, we could not prevent such third party from marketing ondansetron for AUD at those doses.
Results from clinical studies suggest that high intravenous doses of ondansetron may affect the electrical activity of the heart. In a Drug Safety Communication dated June 29, 2012, the FDA stated that: “A 32 mg single intravenous dose of ondansetron (Zofran, ondansetron hydrochloride, and generics) may affect the electrical activity of the heart (QT interval prolongation), which could pre-dispose patients to develop an abnormal and potentially fatal heart rhythm known as Torsades de Pointes.” In addition: “No single intravenous dose should exceed 16 mg.” There are also several recent lawsuits claiming that Zofran® used for the unapproved use of morning sickness causes birth defects. Although we do not believe that our dosage will cause such adverse event there can be no assurance that the negative side effects of the generic drug that have been found in higher dosages will not occur in our dosage or otherwise deter potential users of our product candidate and adversely impact sales of our product candidate. If we were to be required to have such a warning on our drug label, patients may be deterred from using our product candidates.
In addition, we also face the risk, that doctors will prescribe off label, the generic form of ondansetron to treat AUD despite the different dosage of ondansetron in the generic form from that in AD04, the lack of demonstrated clinical efficacy against AUD at the currently available doses (i.e., the Zofran ® and approved generics), and the potential safety concerns if the currently available/higher doses are taken chronically as would be needed for AUD or other addictions. Physicians, or their patients, could divide the lowest dose existing oral tablet into more than ten parts to approximate the necessary AD04 dosage.
Although we believe that any attempt by competitors to reformulate and market ondansetron at our intended dosage levels, while technically feasible, infringes on our intellectual property rights, and should, accordingly, be actionable, we cannot give assurances that we would be successful in defending our rights or that we will have access to sufficient funds necessary to successfully prosecute any such violations of, or infringements on, our intellectual property rights. Additionally, we cannot ensure investors that other companies will not discover and seek to commercialize low doses of ondansetron, not currently available, for other indications.
Coronavirus could adversely impact our business, including our clinical trials.
In December 2019, a novel strain of coronavirus, COVID-19, was reported to have surfaced in Wuhan, China. Since then, the COVID-19 coronavirus has spread to multiple countries, including countries in Europe which we have planned or active clinical trial sites. As the COVID-19 coronavirus continues to spread around the globe, we will likely experience disruptions that could severely impact our business and clinical trials, including:
| | ● | delays or difficulties in enrolling patients in our clinical trials; |
| | ● | delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff; |
| | ● | diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; |
| | ● | interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others; |
| | ● | limitations in employee resources that would otherwise be focused on the conduct of our clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people; |
| | ● | delays in receiving approval from local regulatory authorities to initiate our planned clinical trials; |
| | ● | delays in clinical sites receiving the supplies and materials needed to conduct our clinical trials; |
| | ● | interruption in global shipping that may affect the transport of clinical trial materials, such as investigational drug product used in our clinical trials; |
| | ● | changes in local regulations as part of a response to the COVID-19 coronavirus outbreak which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether; |
| | ● | delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; |
| | ● | delay in the timing of interactions with the FDA due to absenteeism by federal employees or by the diversion of their efforts and attention to approval of other therapeutics or other activities related to COVID-19; and |
| | ● | refusal of the FDA to accept data from clinical trials in affected geographies outside the United States. |
In addition, the outbreak of the coronavirus (“COVID-19”) could continue to disrupt our operations due to absenteeism by infected or ill members of management or other employees, or absenteeism by members of management and other employees who elect not to come to work due to the illness affecting others in our office or laboratory facilities, or due to quarantines. COVID-19 illness could also impact members of our Board of Directors resulting in absenteeism from meetings of the directors or committees of directors, and making it more difficult to convene the quorums of the full Board of Directors or its committees needed to conduct meetings for the management of our affairs.
The global outbreak of the COVID-19 coronavirus continues to rapidly evolve. The extent to which the COVID-19 coronavirus may impact our business and clinical trials will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread and possible resurgences of the disease, the duration of the outbreak, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States and other countries to contain and treat the disease.
While there exists a large body of evidence supporting the safety of our primary API, ondansetron, under short-term use, there are currently no long-term use clinical safety data available.
We intend to market our products, particularly AD04, for long-term use by patients seeking to reduce their number of days of heavy drinking, and we assume future sales volumes reflecting such extended use.
Studies of Zofran® conducted as part of its FDA and other regulatory agencies review process found that the drug is well-tolerated and results in few adverse side effects at dosages almost 100 times the dosage expected to be formulated in AD04. However, to the best of our knowledge, no comprehensive clinical study has been performed to date that has evaluated the safety profile of ondansetron for long-term use. We expect the FDA will require us to provide safety data in at least 100 patients for 12 months and can offer no assurances that safety results of these long term use studies will lead to any subsequent approval for long-term use. There can be no assurance that long-term usage of ondansetron, at dosages anticipated by us, will be safe. Though the FDA has stated it will not require additional non-clinical testing nor will it require a QT interval prolongation clinical study, such statements by the FDA are not legally binding on the agency.
All of our current data for our lead product candidate are the result of Phase 2 clinical trials conducted by third parties and do not necessarily provide sufficient evidence that our products are viable as potential pharmaceutical products.
Through our proprietary access to relevant laboratory and clinical trial results of the University of Virginia’s research program, and through our reliance on publicly available third-party research, we possess toxicology, pharmacokinetic, and other preclinical data and clinical data on AD04. As of now, AD04 has completed only Phase 2 clinical trials and we are now conducting its first Phase 3 trial. There is no guarantee that Phase 2 results can or will be replicated by pivotal Phase 3 studies.
To date, long-term safety and efficacy have not yet been demonstrated in clinical trials for our investigational product candidate. Favorable results in early studies or trials may not be repeated in later studies or trials. Even if our clinical trials are initiated and completed as planned, we cannot be certain that the results will support our product candidate claims. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. We cannot be sure that the results of later clinical trials would replicate the results of prior clinical trials and preclinical testing, nor that they would satisfy the requirements of the FDA or other regulatory agencies. Clinical trials may fail to demonstrate that our product candidate is safe for humans and effective for indicated uses. Preclinical and clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization. Any delay in, or termination of, our clinical trials would delay our obtaining FDA or EMA approval for the affected product candidate and, ultimately, our ability to commercialize that product candidate.
Previous clinical trials using ondansetron have had different trial designs, doses, parameters and endpoints than the current ONWARD Phase 3 clinical trial that is expected to serve as a basis for approval of AD04. Though various doses of ondansetron have been tested as treatments for alcohol addiction (Johnson, BA et al., 2011; Johnson, BA et al., 2000; Kranzler et al, 2003; Sellers, EM et al., 1994), the 283-patient Phase 2b clinical trial on which we are largely basing our clinical expectations only tested one dosing regimen, which was weight-based (Johnson, BA et al., 2011). We plan to use a fixed dose in future clinical trials that we believe provides good coverage given the dose ranges tested clinically; however, it is possible that the dose selected will not be the optimal dose and so drug effects may be limited or not be demonstrated sufficiently in clinical testing. Additionally, only one genotype in the genetic panel that will be used to define patients that are genotype positive for treatment with AD04 was used in primary analyses of the Phase 2b trial and three of the genotypes were added to the panel after a retrospective exploratory analysis of the Phase 2b data. The genotype in the panel related to the 5-HTT, that was included in the primary analysis (Johnson, BA et al., 2011) appears to make up about half of the patients that are genotype positive. The three genotypes related to modulation of the 5-HT3 receptor were selected based on a retrospective analysis that was constrained to 18 single-nucleotide polymorphism (“SNPs”) identified for analysis (Johnson, BA et al., 2013). Therefore, confidence in the effects of the 5-HT3 genetics is less than that for the 5-HTT genetics, and this could negatively impact the treatment effect of AD04 in Phase 3 trials for a segment of the patients identified as genotype positive, which could dilute the overall demonstrated effect of AD04 in the trial.
The endpoints for the Phase 2b clinical trial of AD04 were reduction in the severity of drinking, measured as drinks per day of drinking alcohol and reduction frequency of drinking, measured by days of total abstinence from alcohol. These are surrogate endpoints for the endpoints expected to be required for approval, which, for Europe, are expected to be reduction of heavy drinking days (defined herein), measured in percentage of heavy drinking days per month, and total average alcohol consumed per month, and, for the United States, is expected to be the percentage of patients that have no heavy drinking days in the final 2 months of a six month treatment regimen of AD04. Though the Phase 2b trial showed a statistically significant effect against both pre-specified endpoints and when analyzed for reducing heavy drinking days, all when compared against the placebo group, it is possible that AD04 could affect the endpoints of the Phase 2b trial while not demonstrating a strong enough effect to gain approval.
The Phase 2b clinical trial was 12 weeks in duration, including a one week placebo run-in period, and the Phase 3 trials expected to be required for approval will be 24 weeks. Though the effect of AD04 against AUD in the Phase 2b trial appeared to begin in the first month of the trial and appeared durable throughout the trial, we cannot be sure the effect will extend for the duration of the Phase 3 trials.
The FDA and/or EMA may not accept our planned Phase 3 endpoints for final approval of AD04 and may determine additional clinical trials are required for approval of AD04.
The FDA has indicated to us that a comparison of the percent of patients with no heavy drinking days in the last two months of a six month clinical trial between the drug and placebo groups will be a satisfactory endpoint for determination of a successful Phase 3 trial of AD04 and has published the draft guidance Alcoholism: Developing Drugs for Treatment Guidance for Industry dated February 2015 indicating this endpoint for the development of drugs for AUD. Similarly, the EMA has in the past accepted the co-primary endpoints of reduction from baseline in days of heavy drinking and reduction total grams of alcohol consumed per month and has published the Guideline on the development of medicinal products for the treatment of alcohol dependence on February 18, 2010 stating these endpoints as approvable endpoints for alcohol addiction treatment. Despite these indications, neither the FDA nor the EMA is bound to accept the stated endpoint if a new drug application for AD04 is submitted and their definitions of a heavy drinking day may change. We, however, can offer no assurance that the FDA or EMA will approve our primary endpoints, that we can achieve success at the any endpoints they do approve, or that these potential benefits will subsequently be realized.
We will incur additional costs and our approvals could be delayed if the FDA or EMA requires additional clinical trials in patients that are negative for the genotypes targeted by AD04. In addition, clinical trials conducted with only genotype positive subjects will likely result in labeling restricted to treating patients that are genotype positive.
Although the FDA has indicated that it sees little evidence of positive effects for the use of AD04 in subjects that are negative for the genotypes targeted by AD04 and has stated that it would not object to the AD04 Phase 3 clinical trials going forward without including these additional subjects, the FDA has indicated that some research in this area may be required prior to approval of AD04 for AUD within the marker negative population. We believe the data supports our hypothesis that no further studies in genotype negative patients need be conducted. However, the FDA has indicated that any approval based on a trial only in genotype positive subjects would result in labeling restricted to treating patients that are genotype positive. If further studies are required, we will incur additional costs not anticipated, and it could delay approval of AD04 or, if the results of such studies are not positive for AD04, it may result in AD04 not being approved or it may result in AD04’s patents failing to protect AD04 against generic competition.
Under the Pediatric Research Equity Act (“PREA”), NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. We plan to test AD04 in adolescent patients (ages 12-17) as part of our next Phase 3 trial. If successful, we intend to request labeling for treating adolescent patients.
Our use of the currently manufactured clinical trial material in the plan Phase 3 trial is dependent upon the review and approval of the relevant regulatory agencies and authorities.
The Company has manufactured additional clinical trial material for use in the ONWARD trial and other studies that may be required by the FDA or EMA. No assurance can be given that the CMC plan developed by us will be satisfactory to the regulatory agencies or that the clinical trial material produced for use in clinical trials of AD04 will be approved for use in the trials, either of which could result in delay of the clinical trial program and a requirement for increased investment prior to commencement of clinical trials.
Our lead investigational product, AD04, is dependent on a successful development, approval, and commercialization of a genetic test, which is expected to be classified as a companion diagnostic.
Treatment with AD04 will be dependent on identification of patients with a genetic test (i.e., a companion diagnostic). Companion diagnostics and complementary diagnostics are regulated as medical devices by the FDA and, as such, require either clearance or approval prior to commercialization. While the technology for the test we plan to use is well established, it cannot be certain the testing laboratory we set up will be able to conduct the test with the selectivity and sensitivity that will be required or that the genetic test will be approved by FDA for such use, which could increase the time and cost to develop AD04 and possibly prevent marketing approval. While we have been party to a joint meeting with the Center for Drug Evaluation and Research (“CDER”, the FDA division responsible for drug approvals) and the Center for Devices and Radiological Health (“CDRH”, the FDA division responsible for device approvals, including genetic tests) at which agreement was reached as to the development path for the genetic test, neither CDER nor CDRH is bound to accept our planned submission package even if the data is positive. We have been instructed by CDER and CDRH that we need to obtain a separate approval or marketing authorization for the companion diagnostic genetic test from CDRH. We are collecting and storing additional blood samples from all patients enrolled in the ONWARD Phase 3 trial, and plan to do so for any future trials that may be conducted, in the event of any difficulties, however, we cannot be certain we can overcome all of the technological, logistical or regulatory hurdles related to the genetic testing, which include, without limitation, technical validation of the test (e.g. specificity, sensitivity, reproducibility, robustness of methods), clinical validation acceptable to CDER and CDRH, all of which are needed for approval of AD04 and its companion diagnostic genetic test. Failure in any of these areas could delay approval of AD04, increase the cost necessary to achieve approval of AD04 or prevent approval of AD04.
If we obtain approval of AD04 and its genetic test, we currently plan to distribute the genetic test as widely as possible to third party testing companies with limited attention to capitalizing on the revenue potential of the genetic test itself in order to achieve wider availability of the genetic test to drive market uptake of AD04. However, we cannot be sure that third party testing companies will be willing to provide the test, that reimbursement for the test will be available to make such business profitable, or that taking a genetic test will be acceptable to patients or physicians. Additionally, our plans may change so that we attempt to make the test a material business of our own. In this event, the availability of the genetic test in the market could be reduced, limiting market uptake of AD04, the testing business could fail, and we could be in a position where it never reaches profitability. As one of our products/services, the genetic test will be subject to all of the risks stated elsewhere herein related to reimbursement of our products and failure to achieve adequate reimbursement could limit the potential sales of both the genetic test and AD04, and there is no assurance that the diagnostic will be approved or authorized for marketing.
We have limited experience as a company conducting clinical trials.
We are a clinical stage company and our success is dependent upon our ability to obtain regulatory approval for and commercialization of our investigational products, and we have not demonstrated an ability to perform the functions necessary for the approval or successful commercialization of any product candidates. The successful commercialization of any product candidates may require us to perform a variety of functions, including:
| | ● | continuing to undertake preclinical development and successfully enroll patients in clinical trials; |
| | ● | participating in regulatory approval processes; |
| | ● | formulating and manufacturing products; and |
| | ● | conducting sales and marketing activities. |
We have limited experience conducting and enrolling patients in clinical trials. While certain members of our management and staff have significant experience in conducting clinical trials, to date, we have not successfully completed any clinical trials as a company. Until recently, our operations have been limited primarily to organizing and staffing our company, acquiring, developing and securing our proprietary technology and preparing for clinical trials of our product candidate. These operations provide a limited basis to assess our ability to develop and commercialize our product candidate and the advisability of investing in our securities.
All of the preclinical and clinical trials relating to our product candidate have been conducted by third parties. Although we have recruited a team that has significant experience with managing clinical trials, we have no experience as a company in conducting our own clinical trials. In part because of this lack of experience, we cannot guarantee that planned clinical trials will be completed on time, if at all. Large-scale trials require significant additional financial and management resources, monitoring and oversight, and reliance on third-party clinical investigators, contract research organizations (“CROs”), or consultants. Relying on third-party clinical investigators, CROs and manufacturers, which are all also subject to governmental oversight and regulations, may also cause us to encounter delays that are outside of our control.
Our product candidate is in early stages of development.
Because our product candidate is in early stages of development it will require extensive clinical and other testing. Although our lead product candidate has completed a 283-patient Phase 2b clinical trial, we cannot predict with any certainty if or when we might submit an application for regulatory approval for any of our product candidates or whether any such application will be accepted for review by the FDA or EMA, or whether any application will be approved upon review.
Even if our clinical trials are completed as planned, we cannot be certain that their results will support our proposed indications. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and preclinical testing. Results from earlier clinical trials may not be repeated in later clinical trials. The clinical trial process may fail to demonstrate that our product candidate is safe and effective for their proposed uses. This failure could cause us to abandon our product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical trials will delay and possibly preclude the filing of any NDAs with the FDA or EMA and, ultimately, our ability to commercialize our product candidate and generate product revenues.
Our clinical trials may fail to demonstrate adequately the safety and efficacy of AD04 or any future product candidates, which would likely prevent or delay regulatory approval and commercialization.
Before obtaining regulatory approvals for the commercial sale of AD04 or any future product candidates, including AD04, we must demonstrate through lengthy, complex and expensive preclinical testing and clinical trials that product candidates are both safe and effective for use in each target indication. Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of product candidates may not be predictive of the results of later-stage clinical trials. Results from subsequent clinical trials may not be the same as the results from the Phase 2b clinical trial that was conducted by the University of Virginia. There is typically an extremely high rate of attrition from the failure of product candidates proceeding through clinical trials. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy profile despite having progressed through preclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety issues, notwithstanding promising results in earlier trials. We can make no assurances that, should our Phase 3 studies provide statistically significant and clinical meaningful results evidencing that treatment with AD04 results in reduced days of heavy drinking or abstinence, these same results will also provide evidence of greater patient efficacy rates and or patient benefit ratios vis-à-vis currently marketed drug treatments. Most product candidates that commence clinical trials are never approved as products.
In addition, even if the trials are successfully completed, we cannot guarantee that the FDA or foreign regulatory authorities will interpret the results as we do, and more trials could be required before we submit product candidates for approval. To the extent that the results of the trials are not satisfactory to the FDA or foreign regulatory authorities for support of a marketing application, approval of product candidates may be significantly delayed, or we may be required to expend significant additional resources, which may not be available to us, to conduct additional trials in support of potential approval of product candidates.
If we experience delays in the enrollment of patients in our clinical trials our receipt of necessary regulatory approvals could be delayed or prevented.
Although we expect to complete patient enrollment in our landmark ONWARD pivotal Phase 3 clinical trial in the second or third quarter of 2021, our inability to locate and continue to enroll a sufficient number of eligible patients in our current or any future clinical trials would result in significant delays or may require us to abandon one or more clinical trials. Retention of subjects in clinical trials related to AUD can be challenging relative to trials in some other indications due to the nature of the target population. In addition, COVID-19 has made trial operation, including, without limitation, patient enrollment, more difficult and more difficult to project. Our ability to enroll patients in trials is affected by many factors out of our control including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, the prevalence and successful recruiting of patients that are genotype positive, competing clinical trials, and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. Due to the use of a biomarker to determine enrollment in our current and planned Phase 3 clinical trials, we will have a limited population of patients to draw from for our Phase 3 clinical trials.
Global health crises may adversely affect our planned operations.
The conduct of our ongoing ONWARD Phase 3 trial could be materially and adversely affected by the risks, or the public perception of the risks, related to a pandemic or other health crisis, such as the recent outbreak of novel coronavirus (COVID-19). A significant outbreak of contagious diseases in the human population could result in a widespread health crisis that could adversely affect our ongoing trial. Such events could result in the complete or partial closure of one or more of our critical vendors. In addition, an outbreak near our clinical trial site locations would likely impact our ability to recruit patients, delay our clinical trials, and could affect our ability to complete our clinical trials within the planned time periods. Also, public health authorities in the jurisdictions in which our trial is taking place may take steps that would result in significant delay in our trial activities.
Our success will be dependent upon adoption by physicians and others.
Even if the FDA and/or EMA approves our product candidate or any future product candidates we may develop or acquire, the product will require acceptance among physicians, healthcare payers, patients, and the medical community. Our products are to be used in combination with a genetic test targeted at patients with certain specified genotypes. It is anticipated that physicians will recommend patients for screening prior to administration of AD04 or future product candidates. Therefore, our business will be substantially dependent upon our ability to communicate with and obtain support from physicians regarding the benefits of our products relative to alternative treatments available at that time.
Rapid technological change and substantial competition may impair the business.
The pharmaceutical industry is subject to rapid and substantial technological change. Technological competition in the industry from pharmaceutical and biotechnology companies, universities, governmental entities, and others diversifying into the field is intense and is expected to increase. Many of these entities have significantly greater research and development capabilities, as well as substantially more marketing, financial, and managerial resources than we do, and represent significant competition. Acquisitions of, or investments in, competing biotechnology companies by large pharmaceutical companies could increase these competitors’ financial, marketing, and other resources. We cannot assure you that developments by others will not render our products or technologies noncompetitive or that we will be able to keep pace with technological developments. Competitors have developed, or are in the process of developing, technologies that are, or in the future may be, the basis for competitive products. Some of these products may have an entirely different approach or means of accomplishing similar therapeutic endpoints than products we are currently developing. These competing products may be more effective and less costly than the products that we are developing. In addition, conventional behavioral therapies and other treatment approaches currently in use today may continue to be used instead of, rather than in conjunction with, our products.
Any product that we successfully develop, and for which we gain regulatory approval, must compete for market acceptance and market share. Accordingly, important competitive factors, in addition to completion of clinical testing and the receipt of regulatory approval, will include product efficacy, safety, timing, and scope of regulatory approvals, availability of supply, marketing and sales capability, reimbursement coverage, pricing, and patent protection. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. If our products fail to capture and maintain market share, we may not achieve sufficient product revenues and our business will suffer.
We will compete against fully integrated pharmaceutical companies such as Alkermes and Indivior and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors have drugs already approved or in development. In addition, many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs or have substantially greater financial resources than we do, as well as significantly greater experience in:
| | ● | developing drugs, and other therapies; |
| | ● | undertaking preclinical testing and clinical trials; |
| | ● | obtaining FDA and other regulatory approvals of drugs, biologics and other therapies; |
| | ● | formulating and manufacturing drugs, biologics and other therapies; and |
| | ● | launching, marketing and selling drugs, and other therapies. |
Risks Relating to Our Acquisition of Purnovate
The combined company may not experience the anticipated strategic benefits of the Acquisition.
We believe the acquisition of Purnovate will provide certain strategic benefits which would enable Adial to enhance its business and accelerate its business plan through an increased access to capital in the public equity markets. The market price of our common stock may decline as a result of the acquisition if the combined company does not achieve the perceived benefits of the Acquisition as rapidly or to the extent anticipated by us or Purnovate or investors, financial or industry analysts. There can be no assurance that these anticipated benefits of the Acquisition will materialize or that if they materialize will result in increased stockholder value or revenue stream to the combined company.
We may be unable to successfully integrate the Purnovate businesses with its current management and structure.
Our failure to successfully complete the integration of Purnovate could have an adverse effect on our prospects, business activities, cash flow, financial condition, results of operations and stock price. Integration challenges may include the following:
| | ● | assimilating Purnovate’s technology and retaining personnel; |
| | ● | estimating the capital, personnel and equipment required for Purnovate based on the historical experience of management with the businesses they are familiar with; |
| | ● | minimizing potential adverse effects on existing business relationships; and |
| | ● | successfully developing the new products and services. |
Purnovate has had limited operations to date.
Purnovate is a start-up entity and has had limited operations to date. As a start-up entity, Purnovate is subject to many of the risks common to such enterprises, including its ability to implement its business plan, market acceptance of its proposed business and products, under-capitalization, cash shortages, limitations with respect to personnel, financing and other resources, competition from better funded and experienced companies, and uncertainty of its ability to generate revenues. There is no assurance that its activities will be successful or will result in any revenues or profit, and the likelihood of its success must be considered in light of the stage of its development. Even if it generates revenue, there can be no assurance that it will be profitable. In addition, no assurance can be given that it will be able to consummate its business strategy and plans, as described herein, or that financial, technological, market, or other limitations may force it to modify, alter, significantly delay, or significantly impede the implementation of such plans. Purnovate has insufficient results for investors to use to identify historical trends or even to make quarter-to-quarter comparisons of its operating results. Purnovate’s revenue and income potential is unproven and its business model is continually evolving. Purnovate is subject to the risks inherent to the operation of a new business enterprise, and there can be no assurance that Purnovate will be able to successfully address these risks.
Purnovate has a limited operating history upon which to evaluate its ability to commercialize its products.
Purnovate is a development-stage company and its success is dependent upon its ability to develop and commercialize its products and it has not demonstrated an ability to perform the functions necessary for the successful development and commercialization of any product candidates. The successful commercialization of any product candidates will require Purnovate to perform a variety of functions, including:
| | ● | continuing to undertake preclinical development trials and initiating clinical trials; |
| | ● | participating in regulatory approval processes and obtaining regulatory approvals; |
| | ● | formulating and manufacturing products; and |
| | ● | conducting sales and marketing activities. |
Purnovate’s operations have been limited to organizing and staffing Purnovate, acquiring, developing and securing its proprietary technology and undertaking preclinical studies of its product candidates. Purnovate has yet to engage in any clinical trials and therefore the safety of its product candidates is uncertain.
Purnovate’s product candidates are in early stages of clinical trials.
Because Purnovate’s product candidates are in early stages of development they will require extensive preclinical and clinical testing. Purnovate’s lead product has not yet entered clinical trials and cost, speed and ability to advance through clinical trials is uncertain. Purnovate cannot predict with any certainty if or when it might submit an application for regulatory approval for any of its product candidates or whether any such application will be accepted.
Purnovate’s technology may not result in any successful drug candidates.
Purnovate has developed what its believes are lead compounds that could be drug candidates. However, despite there being significant literature and in vitro and in vivo evidence that adenosine analogs may be effective in treating a number of diseases and disorders, the compounds developed to date have not been extensively tested in vitro and have not been tested in vivo. It is possible that any and all compounds or product candidates developed by Purnovate or using its technology may fail or be determined not valuable to pursue as products for a number of reasons, including, without limitation, due to toxicity, lack of efficacy, lack of stability, poor manufacturing characteristics or otherwise.
There is uncertainty as to market acceptance of Purnovate’s technology and products.
Purnovate has conducted its own research into the markets for its products; however, because it will be a new entrant into the market, it cannot guarantee market acceptance of its products and has somewhat limited information on which to estimate anticipated level of sales. Purnovate’s products will require patients and doctors to adopt its technology. Purnovate’s industry is susceptible to rapid technological developments and there can be no assurance that it will be able to match any new technological advances. If it is unable to match the technological changes in the needs of its customers the demand for its products will be reduced.
Risks Relating to Our Business and Industry
If we do not obtain the necessary regulatory approvals in the United States and/or other countries, we will not be able to sell our product candidates.
We cannot assure you that we will receive the approvals necessary to commercialize AD04 or any future product candidates we acquire or develop in the future. We will need FDA approval to commercialize our product candidates in the United States and approvals from the FDA-equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. In order to obtain FDA approval of any product candidate, we must submit to the FDA an NDA, demonstrating that the product candidate is safe, pure and potent, and effective for its intended use. This demonstration requires significant research including preclinical studies, as well as clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. We cannot predict whether our clinical trials will demonstrate the safety and efficacy of our product candidates or if the results of any clinical trials will be sufficient to advance to the next phase of development or for approval from the FDA. We also cannot predict whether our research and clinical approaches will result in drugs or therapeutics that the FDA considers safe and effective for the proposed indications. The FDA has substantial discretion in the approval process.
The approval process may be delayed by changes in government regulation, future legislation or administrative action, or changes in FDA policy that occur prior to or during our regulatory review. Factors that might lead to a suspension or termination of a clinical trial include, but are not limited to:
| | ● | failure to conduct the clinical trial in accordance with U.S., international and or local regulatory requirements; |
| | ● | failure of medical investigators to follow clinical trial protocols; |
| | ● | unforeseen safety issues; and/or |
| | ● | lack of adequate funding to continue the clinical trial. |
| | ● | delays in obtaining regulatory approvals may: |
| | ● | prevent or delay commercialization of, and our ability to derive product revenues from, product candidates; and |
| | ● | diminish any competitive advantages that we may otherwise believe that we hold. |
Even if we comply with all FDA requests, the FDA may ultimately reject one or more of our applications. We may never obtain regulatory clearance for any product candidates. Failure to obtain FDA approval of any of product candidates will severely undermine our business by leaving us without a saleable product, and therefore without any source of revenues, until another product candidate can be developed. There is no guarantee that we will ever be able to develop or acquire another product candidate.
In addition, the FDA may require us to conduct additional preclinical and clinical testing or to perform post-marketing studies, as a condition to granting marketing approval of a product. Initial acceptance by the FDA of clinical trial protocols is subject to constant review and any process control failures could result in additional required testing. Regulatory approval of products often requires that subjects in clinical trials be followed for long periods to assess their overall survival. The results generated after approval could result in loss of marketing approval, changes in product labeling, and/or new or increased concerns about the side effects or efficacy of a product. The FDA has significant post-market authority, including the explicit authority to require post-market studies and clinical trials, labeling changes based on new safety information, and compliance with FDA-approved risk evaluation and mitigation strategies. The FDA’s exercise of its authority has in some cases resulted, and in the future could result, in delays or increased costs during product development, clinical trials and regulatory review, increased costs to comply with additional post-approval regulatory requirements and potential restrictions on sales of approved products based on labeling or other requirements.
In foreign jurisdictions, we must also receive approval from the appropriate regulatory authorities before we can commercialize any candidate products. Foreign regulatory approval processes generally include all of the risks associated with the FDA approval procedures described above. There can be no assurance that we will receive the approvals necessary to commercialize our product candidate for sale outside the United States.
Changes in regulatory requirements and guidance may occur, and we may need to amend clinical trial protocols or our development plan to reflect these changes. Amendments may require resubmitting clinical trial protocols to FDA and institutional review boards for reexamination, which may impact the costs, timing or successful completion of a clinical trial. If we experience delays in completion of, or if we terminate any clinical trials, the commercial prospects for product candidates may be harmed, and the ability to generate product revenues will be delayed. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of product candidates.
Obtaining and maintaining regulatory approval of product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of product candidates in other jurisdictions.
Obtaining and maintaining regulatory approval of product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, and a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including additional preclinical studies or clinical trials, as clinical studies conducted in one jurisdiction may not be accepted by or sufficient for regulatory authorities in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our candidate products is also subject to approval. Additionally, some foreign jurisdictions require participation of subjects from their country in the Phase 3 trials in order to gain approval in their country.
We intend to also submit marketing applications in other jurisdictions, including European countries. Regulatory authorities in jurisdictions outside of the United States have requirements for approval of product candidates with which we must comply prior to marketing in those jurisdictions. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our products in certain countries. If we fail to comply with the regulatory requirements in international markets and/or fail to receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market potential of AD04 or any future product candidates will be harmed.
Even if we receive regulatory approval of AD04 or any future product candidates, we will be subject to ongoing regulatory obligations, such as post market surveillance and current good manufacturing practice (“GMP”) requirements, and continued regulatory review, which may result in significant additional expense. We may also be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with product candidates. In addition, third parties on whom we rely must comply with regulatory requirements, and any non-compliance on their part may negatively impact our business, assuming we obtain regulatory authorization at all.
Any regulatory approvals that we receive for product candidates will require surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require a Risk Evaluation and Mitigation Strategy (“REMS”) program in order to approve product candidates, which could entail requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA could also require a boxed warning, sometimes referred to as a Black Box Warning on the product label to identify a particular safety risk, which could affect commercial efforts to promote and sell the product. In addition, if the FDA or a comparable foreign regulatory authority approves product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for product candidates will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current GMPs and current good clinical practices (“GCPs”) for any clinical trials that we conduct post-approval. We are also subject to certain user fees imposed by the regulatory agencies. Later discovery of previously unknown problems with product candidates, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| | ● | restrictions on the marketing or manufacturing of product candidates, withdrawal of the product from the market, or product recalls; |
| | ● | fines, warning letters or holds on clinical trials; |
| | ● | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or suspension or revocation of approvals; |
| | ● | product seizure or detention, or refusal to permit the import or export of product candidates; and |
| | ● | injunctions or the imposition of civil or criminal penalties. |
The FDA’s and other regulatory authorities’ policies may change, such as those required by the 21st Century Cures Act, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of AD04 or any future product candidates. In addition, it is unclear what changes, if any, the new presidential administration may bring. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
Clinical trials are very expensive, time-consuming and difficult to design and implement.
As part of the regulatory process, we must conduct clinical trials for each product candidate to demonstrate safety and efficacy to the satisfaction of the FDA and other regulatory authorities. As we advance AD04 or any future product candidates we expect that our expenses will increase. The number and design of the clinical trials that will be required varies depending upon product candidate, the condition being evaluated, current medical strategies and the trial results themselves. Therefore, it is difficult to accurately estimate the cost of the clinical trials. Clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time consuming. We estimate that clinical trials of product candidates including AD04, will take at least several years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed or prevented by several factors, including:
| | ● | unforeseen safety issues; |
| | ● | failure to determine appropriate dosing; |
| | ● | greater than anticipated cost of our clinical trials; |
| | ● | failure to demonstrate effectiveness during clinical trials; |
| | ● | slower than expected rates of subject recruitment or difficulty obtaining investigators; |
| | ● | subject drop-out or discontinuation; |
| | ● | inability to monitor subjects adequately during or after treatment; |
| | ● | third party contractors, including, without limitation, CRO’s and manufacturers, failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; |
| | ● | reaching agreements with prospective CROs, and trial sites, both of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| | ● | insufficient or inadequate supply or quality of product candidates or other necessary materials to conduct our trials; |
| | ● | potential additional safety monitoring, or other conditions required by FDA or comparable foreign regulatory authorities regarding the scope or design of our clinical trials, or other studies requested by regulatory agencies; |
| | ● | problems engaging Institutional Review Boards (“IRBs”), to oversee trials or in obtaining and maintaining IRB approval of studies; |
| | ● | imposition of clinical hold or suspension of our clinical trials by regulatory authorities; and |
| | ● | inability or unwillingness of medical investigators to follow our clinical protocols. |
In addition, we or the FDA may suspend or terminate our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our Investigational New Drug, or IND, submissions or the conduct of these trials. Therefore, we cannot predict with any certainty when, if ever, future clinical trials will commence or be completed.
AD04 and any future product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential or result in significant negative consequences.
Undesirable side effects caused by AD04 or any future product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign regulatory authorities. Results of our trials could reveal a high and unacceptable severity and prevalence of side effects or other unexpected characteristics.
If unacceptable safety concerns or other adverse events arise in the development of a product candidate, our clinical trials could be suspended or terminated or the FDA or comparable foreign regulatory authorities could order us to cease clinical trials or deny approval of such product candidate for any or all targeted indications. Treatment-related side effects could also affect patient recruitment or the ability of enrolled subjects to complete the trial or result in potential product liability claims. Inadequate training in recognizing or managing the potential side effects of a product candidate could result in patient deaths. Any of these occurrences may harm our business, financial condition and prospects significantly.
We may incur substantial liabilities and may be required to limit commercialization of our products in response to product liability lawsuits.
The testing and marketing of drug product candidates entail an inherent risk of product liability. Product liability claims might be brought against us by consumers, health care providers or others selling or otherwise coming into contact with our products. Clinical trial liability claims may be filed against us for damages suffered by clinical trial subjects or their families. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products which could impact our ability to continue as a going concern. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with collaborators. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| | ● | decreased demand for any approved product candidates; |
| | ● | impairment of our business reputation; |
| | ● | withdrawal of clinical trial participants; |
| | ● | costs of related litigation; |
| | | |
| | ● | distraction of management’s attention; |
| | ● | substantial monetary awards to patients or other claimants; |
| | ● | the inability to successfully commercialize any approved drug candidates. |
There is uncertainty as to market acceptance of our technology and product candidates.
Even if the FDA approves our current product candidate, or any future product candidates we may develop or acquire, the products may not gain broad market acceptance among physicians, healthcare payers, patients, and the medical community. We have conducted our own research into the markets for our product candidates; however, we cannot guarantee market acceptance of our product candidates, if approved, and have somewhat limited information on which to estimate our anticipated level of sales. Product candidates, if approved, will require patients, healthcare providers and doctors to adopt our technology. Our industry is susceptible to rapid technological developments and there can be no assurance that we will be able to match any new technological advances. If we are unable to match the technological changes in the needs of our customers, the demand for our products will be reduced. Acceptance and use of any products we market, assuming market authorization approval at all, will depend upon a number of factors including:
| | ● | perceptions by members of the health care community, including physicians, about the safety and effectiveness of our products; |
| | ● | limitation on use or warnings required by FDA in our product labeling; |
| | ● | cost-effectiveness of our products relative to competing products; |
| | ● | convenience and ease of administration; |
| | ● | potential advantages of alternative treatment methods; |
| | ● | availability of reimbursement for our products from government or other healthcare payers; and |
| | ● | effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any. |
Because we expect virtually all of our product revenues for the foreseeable future to be generated from sales of AD04, if approved, the failure of this product to find market acceptance would substantially harm our business and would adversely affect our revenue.
Even if we are able to obtain regulatory approval for our product candidate or any product candidates we develop or acquire, we will continue to be subject to ongoing and extensive regulatory requirements, and our failure, or the failure of our contract manufacturers, to comply with these requirements could substantially harm our business.
If the FDA approves our product candidate or any product candidates we develop or acquire, the labeling, manufacturing, packaging, adverse events reporting, storage, advertising, promotion and record-keeping for our products will be subject to ongoing FDA requirements and continued regulatory oversight and review. We may also be subject to additional FDA post-marketing obligations. If we are not able to maintain regulatory compliance, we may not be permitted to market product candidates and/or may be subject to product recalls or seizures. The subsequent discovery of previously unknown problems with any marketed product, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the product, and could include withdrawal of the product from the market.
Our employees, independent contractors, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee fraud or other illegal activity by our employees, independent contractors, consultants, commercial partners and vendors. Misconduct by these parties could include intentional, reckless and/or negligent conduct that fails to: (i) comply with the laws of the FDA and other similar foreign regulatory bodies; (ii) provide true, complete and accurate information to the FDA and other similar foreign regulatory bodies; (iii) comply with manufacturing standards we have established; (iv) comply with healthcare fraud and abuse laws in the United States and similar foreign fraudulent misconduct laws; or (v) report financial information or data accurately or to disclose unauthorized activities to us. Any such misconduct or noncompliance could negatively affect the FDA’s review of our regulatory submission, including delaying approval or disallowance of certain information to support the submission, and/or delay a federal or state healthcare program’s or a commercial insurer’s determination regarding the availability of future reimbursement for product candidates. If we obtain FDA approval of any product candidates and begin commercializing those products in the United States, our potential exposure under such laws will increase significantly, and our costs associated with compliance with such laws are also likely to increase. These laws may impact, among other things, our current activities with principal investigators and research patients, as well as proposed and future sales, marketing and education programs. In particular, the promotion, sales and marketing of healthcare items and services, as well as certain business arrangements in the healthcare industry, are subject to extensive laws designed to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, structuring and commission(s), certain customer incentive programs and other business arrangements generally. Activities subject to these laws also involve the improper use of information obtained in the course of patient recruitment for clinical trials. The laws that may affect our ability to operate or may require us to modify certain programs include, but are not limited to:
| | ● | the federal Anti-Kickback Statute, which prohibits, among other things, knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for, either the referral of an individual, or the purchase, lease, order or recommendation of any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs; |
| | ● | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid, or other third-party payors (both governmental and private) that are false or fraudulent or knowingly making a false statement to improperly avoid, decrease or conceal an obligation to pay money to a federal or state healthcare program or private payor; |
| | ● | the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which, among other things, created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters; |
| | ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (“HITECH”), and their respective implementing regulations, which, among other things, impose requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform services for them that involve the use, or disclosure of, individually identifiable health information, relating to the privacy, security and transmission of such individually identifiable health information; |
| | ● | the federal Physician Payment Sunshine Act, created under the Healthcare Reform Act (as defined herein), and its implementing regulations, which require certain manufacturers of drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the United States Department of Health and Human Services (“HHS”), information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; |
| | ● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
| | ● | the Foreign Corrupt Practices Act (the “FCPA”) and similar antibribery and anticorruption laws in other countries that, for example, prevent improper payments or transfers of anything of value to foreign officials for the purpose of gaining commercial advantage, obtaining or retaining business, or to enhancing clinical trials. |
Additionally, we are subject to state and foreign equivalents of each of the healthcare laws described above, among others, some of which may be broader in scope and may apply regardless of the payor.
It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent inappropriate conduct may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Efforts to ensure that our business arrangements will comply with applicable healthcare laws may involve substantial costs. It is possible that governmental and enforcement authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations. In addition, the approval and commercialization of any product candidates outside the United States will also likely subject us to foreign equivalents of the healthcare laws mentioned above, among other foreign laws.
We have no experience selling, marketing or distributing products and have no internal capability to do so.
We currently have no sales, marketing or distribution capabilities, including, without limitation, capabilities to market AD04 or its companion genetic test. We do not anticipate having the resources in the foreseeable future to allocate to the sales and marketing of our proposed products, if approved. Our future success depends, in part, on our ability to enter into and maintain collaborative relationships for such capabilities, the collaborator’s strategic interest in the products under development and such collaborator’s ability to successfully market and sell any such products. We intend to pursue collaborative arrangements regarding the sales and marketing of our products, however, there can be no assurance that we will be able to establish or maintain such collaborative arrangements, or if able to do so, that our collaborators will have effective sales forces. To the extent that we decide not to, or are unable to, enter into collaborative arrangements with respect to the sales and marketing of our proposed products, significant capital expenditures, management resources and time will be required to establish and develop an in-house marketing and sales force with technical expertise. There can also be no assurance that we will be able to establish or maintain relationships with third party collaborators or develop in-house sales and distribution capabilities. To the extent that we depend on third parties for marketing and distribution, any revenues we receive will depend upon the efforts of such third parties over whom we have no control, and there can be no assurance that such efforts will be successful. In addition, there can also be no assurance that we will be able to successfully market and sell our products in the United States or overseas on our own.
We may not be successful in establishing and maintaining strategic partnerships, which could adversely affect our ability to develop and commercialize products.
We may seek to enter into strategic partnerships in the future, including alliances with other biotechnology or pharmaceutical companies, to enhance and accelerate the development and commercialization of our products, such as a third party drug development company. We face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex and can be costly. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for any future product candidates and programs because our research and development pipeline may be insufficient, our product candidates and programs may be deemed to be at too early of a stage of development for collaborative effort and/or third parties may not view our product candidates and programs as having the requisite potential to demonstrate safety and efficacy or return on investment. Even if we are successful in our efforts to establish strategic partnerships, the terms that we agree upon may not be favorable to us and we may not be able to maintain such strategic partnerships if, for example, development or approval of a product candidate is delayed or sales of an approved product are disappointing.
If we ultimately determine that entering into strategic partnerships is in our best interest but either fail to enter into, are delayed in entering into or fail to maintain such strategic partnerships:
| | ● | the development of our current product candidate or certain future product candidates may be terminated or delayed; |
| | ● | our planned clinical trials may be restructured or terminated; |
| | ● | our cash expenditures related to development of our current product candidate or certain future product candidates may increase significantly and we may need to seek additional financing; |
| | ● | we may be required to hire additional employees or otherwise develop expertise, such as sales and marketing expertise, for which we have not budgeted; |
| | ● | we will bear all of the risk related to the development of any such product candidates; and |
| | ● | the competitiveness of any product candidate that is commercialized could be reduced. |
To the extent we elect to enter into licensing or collaboration agreements to partner AD04 or any future product candidates, our dependence on such relationships may adversely affect our business.
Our commercialization strategy for certain product candidates may depend on our ability to enter into agreements with collaborators to obtain assistance and funding for the development and potential commercialization of these investigational product candidates. Supporting diligence activities conducted by potential collaborators and negotiating the financial and other terms of a collaboration agreement are long and complex processes with uncertain results. Even if we are successful in entering into one or more collaboration agreements, collaborations may involve greater uncertainty for us, as we have less control over certain aspects of our collaborative programs than we do over our proprietary development and commercialization programs. Our collaborators could delay or terminate their agreements, and our product candidates subject to collaborative arrangements may never be successfully developed or commercialized.
Further, our future collaborators may develop alternative products or pursue alternative technologies either on their own or in collaboration with others, including our competitors, and the priorities or focus of our collaborators may shift such that our programs receive less attention or fewer resources than we would like, or they may be terminated altogether. Any such actions by our collaborators may adversely affect our business prospects and ability to earn revenues. In addition, we could have disputes with our future collaborators, such as the interpretation of terms in our agreements. Any such disagreements could lead to delays in the development or commercialization of any potential products or could result in time-consuming and expensive litigation or arbitration, which may not be resolved in our favor.
Our internal computer systems, or those used by our CROs or other contractors or consultants, may fail or suffer security breaches.
Despite the implementation of security measures, our internal computer systems and those of our future CROs and other contractors and consultants are vulnerable to damage from computer viruses and unauthorized access. While we have not experienced any such material system failure or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Since we rely on third parties for research and development of AD04 and expect do so for future product candidates and for the manufacture of product candidates and to conduct clinical trials, similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of product candidates could be delayed.
We have limited protection for our intellectual property. Our licensed patents and proprietary rights may not prevent us from infringing on the rights of others or prohibit potential competitors from commercializing products.
We intend to rely on a combination of common law copyright, patent, trademark, and trade secret laws and measures to protect our proprietary information. We have licensed patents to protect certain of our proprietary intellectual property and have obtained exclusive rights to license certain of the technology for which patent protection has been obtained; however, such protection does not prevent unauthorized use of such technology. Trademark and copyright protections may be limited, and enforcement could be too costly to be effective. It may also be possible for unauthorized third parties to copy aspects of, or otherwise obtain and use, our proprietary information without authorization, including, but not limited to, product design, software, customer and prospective customer lists, trade secrets, copyrights, patents and other proprietary rights and materials. Other parties can use and register confusingly similar business, product and service names, as well as domain names, which could divert customers, resulting in a material adverse effect on our business, operating results and financial condition.
We have not conducted an exhaustive patent search and cannot assure you that patents do not exist or could not be filed that would negatively affect our ability to market our products or maintain our competitive position with respect to our products. Additionally, our licensed patents may not prevent others from developing competitive products using related technology. Furthermore, other companies that obtain patents claiming products or processes useful to us may bring infringement actions against us. As a result, we may be required to obtain licenses from others to develop, manufacture or market our products. We cannot assure you that we will be able to obtain any such licenses on commercially reasonable terms, if at all.
We also rely on trade secrets and proprietary know-how that we seek to protect, in part, by confidentiality agreements with our employees, consultants, suppliers, and licensees. We cannot give any assurance that these third parties will not breach these agreements, that we would have adequate remedies for any breach, or that our trade secrets will not otherwise become known or be independently developed by competitors.
We cannot assure you that the U.S. Patent and Trademark Office (“USPTO”) will approve pending patent applications for intellectual property for which we are currently the exclusive worldwide licensee, or that any patent issued to, or licensed by, us will provide protection that has commercial significance. In this regard, the patent position of pharmaceutical compounds and compositions is particularly uncertain. Even issued patents may later be modified or revoked by the USPTO in proceedings instituted by others or by us. In addition, we cannot assure you that our licensed patents will afford protection against competitors with similar compounds or technologies, that others will not obtain patents with claims similar to those covered by our licensed patents or applications, or that the patents of others will not adversely affect our ability to conduct our business.
Despite licensing patents issued in more than 40 jurisdictions around the world, continuing to achieve additional foreign patent issuances and maintaining and defending foreign patents may be more difficult than defending domestic patents because of differences in patent laws, and our licensed patent position therefore may be stronger in the United States than abroad. In addition, the protection provided by foreign patents, once they are obtained, may be weaker than that provided in the United States.
If we fail to successfully enforce our intellectual property rights, our competitive position could suffer, which could harm our operating results. Competitors may challenge the validity or scope of our licensed patents or future patents we may obtain or license. In addition, our licensed patents may not provide us with a meaningful competitive advantage. We may be required to spend significant resources to monitor and police our licensed intellectual property rights. We may not be able to detect infringement and our competitive position may be harmed. In addition, competitors may design around our technology or develop competing technologies. Intellectual property rights may also be unavailable or limited in some foreign countries, which could make it easier for competitors to capture market share.
The technology we license, our products or our development efforts may be found to infringe upon third-party intellectual property rights.
Our commercial success depends in part on us avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation involving patents and other intellectual property rights in the biotechnology and pharmaceutical industries, as well as administrative proceedings for challenging patents, including interference and reexamination proceedings before the USPTO, or oppositions and other comparable proceedings in other jurisdictions. Recently, under the American Invents Act (“AIA”), new procedures including inter parties review and post grant review have been implemented. These procedures are relatively new and the manner in which they are being implemented continues to evolve, which brings additional uncertainty to our licensed patents and pending applications. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates may give rise to claims of infringement of the patent rights of others.
Third parties may, in the future, assert claims or initiate litigation related to their patent, copyright, trademark and other intellectual property rights in technology that is important to us. The asserted claims and/or litigation could include claims against us, our licensors or our suppliers alleging infringement of intellectual property rights with respect to our products or components of those products. Regardless of the merit of the claims, they could be time consuming, result in costly litigation and diversion of technical and management personnel, or require us to develop a non-infringing technology or enter into license agreements. We have not undertaken an exhaustive search to discover any third party intellectual patent rights which might be infringed by commercialization of the product candidates described herein. Although we are not currently aware of any such third party intellectual patent rights, it is possible that such rights currently exist or might be obtained in the future. In the event that a third party controls such rights and we are unable to obtain a license to such rights on commercially reasonable terms, we may not be able to sell or continue to develop our products, and may be liable for damages for such infringement. We cannot assure you that licenses will be available on acceptable terms, if at all. Furthermore, because of the potential for significant damage awards, which are not necessarily predictable, it is not unusual to find even arguably unmeritorious claims resulting in large settlements. If any infringement or other intellectual property claim made against us by any third party is successful, or if we fail to develop non-infringing technology or license the proprietary rights on commercially reasonable terms and conditions, our business, operating results and financial condition could be materially adversely affected.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to:
| | ● | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
| | ● | abandon an infringing drug or therapy candidate; |
| | ● | redesign our products or processes to avoid infringement; |
| | ● | stop using the subject matter claimed in the patents held by others; |
| | ● | defend litigation or administrative proceedings which may be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources. |
Parties making claims against us may seek and obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible or require substantial time and monetary expenditure. We cannot predict whether any such license would be available at all or whether it would be available on commercially reasonable terms. Furthermore, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of product candidates. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to further develop and commercialize product candidates, which could harm our business significantly.
We may be involved in lawsuits to protect or enforce the patents of our licensors, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that one or more of our licensed patents is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our licensed patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our licensed patents at risk of being invalidated, held unenforceable, or interpreted narrowly and could put our licensed patent applications at risk of not issuing. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible or require substantial time and monetary expenditure.
Interference proceedings provoked by third parties or brought by the USPTO may be necessary to determine the priority of inventions with respect to some of our licensed patents or patent applications subject to pre-AIA or those of our licensors. An unfavorable outcome could result in a loss of our current licensed patent rights and could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Litigation or interference proceedings may result in a decision adverse to our interests and, even if we are successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
A derivation proceeding is a trial proceeding conducted at the Patent Trial and Appeal Board to determine whether (i) an inventor named in an earlier application derived the claimed invention from an inventor named in the petitioner’s application; and (ii) the earlier application claiming such invention was filed without authorization. An applicant subject to the first-inventor-to-file provisions may file a petition to institute a derivation proceeding only within one year of the first publication of a claim to an invention that is the same or substantially the same as the earlier application’s claim to the invention. The petition must be supported by substantial evidence that the claimed invention was derived from an inventor named in the petitioner’s application. Derivation proceedings may result in a decision adverse to our interests and, even if we are successful, may result in substantial costs and distract our management and other employees.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our shares of common stock.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Noncompliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In such an event, our competitors might be able to enter the market, which would have a material adverse effect on our business.
Patents are subject to changing legal interpretation by the USPTO and the Courts.
If the U.S. Supreme Court, other federal courts, or the USPTO were to change the standards of patentability such changes could have a negative impact on our business. Recent court cases have made it more difficult to protect certain types of inventions. For instance, on October 30, 2008, the Court of Appeals for the Federal Circuit issued a decision that methods or processes cannot be patented unless they are tied to a machine or involve a physical transformation. On March 20, 2012, in the case Mayo v. Prometheus, the U.S. Supreme Court invalidated a patent focused on a diagnostic process because the patent claim embodied a law of nature. On July 3, 2012, the USPTO issued its Interim Guidelines for Subject Matter Eligibility Analysis of Process Claims Involving Laws of Nature in view of the Prometheus decision. It remains to be seen how these guidelines will play out in the actual prosecution of diagnostic claims. Similarly, it remains to be seen how lower courts will interpret the Prometheus decision. Some aspects of our technology involve processes that may be subject to this evolving standard and we cannot guarantee that any of our pending process claims will be patentable as a result of such evolving standards.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial cost and be a distraction to our management and employees.
Our ability to generate product revenues will be diminished if our products sell for inadequate prices or patients are unable to obtain adequate levels of reimbursement.
Our ability to commercialize our products, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from:
| | ● | government and health administration authorities; |
| | ● | private health maintenance organizations and health insurers; and |
| | ● | other healthcare payers. |
Patients generally expect that products such as ours are covered and reimbursed by third-party payors for all or part of the costs and fees associated with their use. If such products are not covered and reimbursed then patients may be responsible for the entire cost of the product, which can be substantial. Therefore, health care providers generally do not prescribe products that are not covered and reimbursed by third-party payors in order to avoid subjecting their patients to such financial liability. The existence of adequate coverage and reimbursement for the products by government and private insurance plans is central to the acceptance of AD04 and any future products we provide.
During the past several years, third-party payors have undertaken cost-containment initiatives including different payment methods, monitoring health care expenditures, and anti-fraud initiatives. For some governmental programs, such as Medicaid, coverage and reimbursement differ from state to state, and some state Medicaid programs may not pay an adequate amount for AD04 or any of our other products or may make no payment at all. Furthermore, the health care industry in the United States has experienced a trend toward cost containment as government and private insurers seek to control health care costs by imposing lower payment rates and negotiating reduced contract rates with service providers. Therefore, we cannot be certain that our services will be reimbursed at a level that is sufficient to meet our costs.
Obtaining coverage and reimbursement approval of a product from a government or other third-party payor is a time-consuming and costly process that could require us to provide to the payor supporting scientific, clinical and cost-effectiveness data for the use of our products. Even if we obtain coverage for a given product, the resulting reimbursement payment rates might not be adequate for us to achieve or sustain profitability or may require co-payments that patients find unacceptably high. Patients are unlikely to use AD04 or any future product candidates unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of AD04 or any future product candidates.
We intend to seek approval to market AD04 and future product candidates in both the United States and in selected foreign jurisdictions. If we obtain approval in one or more foreign jurisdictions for AD04 or any future product candidates, we will be subject to rules and regulations in those jurisdictions. In some foreign countries, particularly those in the European Union, the pricing of drugs is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after obtaining marketing approval of a product candidate. In addition, market acceptance and sales of product candidates will depend significantly on the availability of adequate coverage and reimbursement from third-party payors for product candidates and may be affected by existing and future health care reform measures.
Third-party payors, whether domestic or foreign, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could impact our ability to sell our products profitably. In particular, in 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the “Healthcare Reform Act”), was enacted. The Healthcare Reform Act and its implementing regulations, among other things, revised the methodology by which rebates owed by manufacturers to the state and federal government for covered outpatient drugs, including product candidates, under the Medicaid Drug Rebate Program are calculated, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations, subjected manufacturers to new annual fees and taxes for certain branded prescription drugs, and provided incentives to programs that increase the federal government’s comparative effectiveness research.
Other legislative changes have been proposed and adopted in the United States since the Healthcare Reform Act was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers up to 2% per fiscal year. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012 (the “ATRA”) which delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. In March 2013, the President signed an executive order implementing sequestration, and in April 2013, the 2% Medicare payment reductions went into effect. The ATRA also, among other things, reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future, particularly in light of the new presidential administration in the United States, and any proposed changes to healthcare laws that could potentially affect our clinical development or regulatory strategy. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
| | ● | the demand for AD04, or future product candidates, if we obtain regulatory approval; |
| | ● | our ability to set a price that we believe is fair for our products; |
| | ● | our ability to generate revenue and achieve or maintain profitability; |
| | ● | the level of taxes that we are required to pay; and |
| | ● | the availability of capital. |
Any reduction in reimbursement from Medicare, Medicaid or other government programs may result in a similar reduction in payments from private payors, which may adversely affect our future profitability.
If we are unable to obtain adequate coverage and reimbursement for our tests, it is unlikely that our tests will gain widespread acceptance.
Use of our product candidate will require pre-treatment screening. Our strategy for AD04 aims to integrate pre-treatment screening into the drug label, effectively creating a patient-specific or “precision” treatment into one integrated therapeutic offering. Our ability to generate revenue will depend upon the availability of adequate coverage and reimbursement for our tests from third-party payors, including government programs such as Medicare and Medicaid, private insurance plans and managed care programs. Health care providers that order diagnostic services generally expect that those diagnostic services are covered and reimbursed by third-party payors for all or part of the costs and fees associated with the diagnostic tests they order. If such diagnostic tests are not covered and reimbursed then their patients may be responsible for the entire cost of the test, which can be substantial. Therefore, health care providers generally do not order tests that are not covered and reimbursed by third-party payors in order to avoid subjecting their patients to such financial liability. The existence of adequate coverage and reimbursement for the procedures performed by us by government and private insurance plans is central to the acceptance of our product candidate. During the past several years, third-party payors have undertaken cost-containment initiatives including different payment methods, monitoring health care expenditures, and anti-fraud initiatives. In addition, the Centers for Medicare & Medicaid Services, or CMS, which administers the Medicare program, has taken the position that the algorithm portion of multi-analyst algorithmic assays, or MAAAs, is not a clinical laboratory test and is therefore not reimbursable under the Medicare program. Although this position is only applicable to tests with a CMS determined national payment amount, it is possible that the local MACs, who make coverage and payment determinations for tests such as ours may adopt this policy and reduce payment for such test. If that were to happen, reimbursement for our pre-screening tests would be uncertain. We may not be able to achieve or maintain profitability if third-party payors deny coverage or reduce their current levels of payment, or if our costs of production increase faster than increases in reimbursement levels. Further, many private payors use coverage decisions and payment amounts determined by CMS as guidelines in setting their coverage and reimbursement policies. Future action by CMS or other government agencies may diminish payments to clinical laboratories, physicians, outpatient centers and/or hospitals. Those private payors that do not follow the Medicare guidelines may adopt different coverage and reimbursement policies for us and coverage and the amount of reimbursement under those polices is uncertain. For some governmental programs, such as Medicaid, coverage and reimbursement differ from state to state, and some state Medicaid programs may not pay an adequate amount for MyPRS® or may make no payment at all. As the portion of the U.S. population over the age of 65 and eligible for Medicare continues to grow, we may be more vulnerable to coverage and reimbursement limitations imposed by CMS. Furthermore, the health care industry in the United States has experienced a general trend toward cost containment as government and private insurers seek to control health care costs through various mechanisms, including imposing limitations on payment rates and negotiating reduced contract rates with service providers, among other things. Therefore, we cannot be certain that our services will be reimbursed at a level that is sufficient to meet our costs.
A variety of risks associated with marketing AD04 or any future product candidates internationally could materially adversely affect our business.
We plan to seek regulatory approval of AD04 and any future product candidates outside of the United States, in particular in European markets, and, accordingly, we expect that we will be subject to additional risks related to operating in foreign countries if we obtain the necessary approvals, including:
| | ● | differing regulatory and reimbursement requirements in foreign countries; |
| | ● | unexpected changes in tariffs, trade barriers, price and exchange controls and other regulatory requirements; |
| | ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| | ● | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
| | ● | foreign taxes, including withholding of payroll taxes; |
| | ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
| | ● | compliance with U.S. and foreign export control regulations, including economic sanctions and embargo programs, each of which may be subject to unexpected changes; |
| | ● | difficulties staffing and managing foreign operations; |
| | ● | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| | ● | potential liability under the FCPA or comparable foreign regulations; |
| | ● | challenges enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States; |
| | ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; |
| | ● | business interruptions resulting from geo-political actions, including war and terrorism; and |
| | ● | potential difficulties that may arise with pharmaceutical company partners under license or other agreement to jointly develop, seek regulatory approval, and commercialize our products. |
These and other risks associated with our international operations may materially adversely affect our ability to attain or maintain profitable operations.
We may not successfully effect our intended expansion.
Our success will depend upon the expansion of our operations and the effective management of our growth, which will place a significant strain on our management and on our administrative, operational and financial resources. To manage this growth, we must expand our facilities, augment our operational, financial and management systems and hire additional qualified personnel. We will need to hire additional qualified personnel with expertise in preclinical and clinical research, government regulation, formulation and manufacturing, sales and marketing and accounting and financing. In particular, over the next 12 months, we expect to hire additional new employees. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success. If we are unable to manage our growth effectively, our business would be harmed.
We rely on key executive officers and scientific, regulatory and medical advisors, and their knowledge of our business and technical expertise would be difficult to replace.
Because of the specialized nature of our business, our ability to maintain a competitive position depends on our ability to attract and retain qualified management and other personnel. We cannot assure you that we will be able to continue to attract or retain such persons.
We are highly dependent on our principal scientific, regulatory and medical advisors and our chief executive officer. We do not have an insurance policy on the life of our chief executive officer, William B. Stilley; and we do not have “key person” life insurance policies for any of our other officers or advisors. The loss of the technical knowledge and management and industry expertise of any of our key personnel could result in delays in product development, loss of customers and sales and diversion of management resources, which could adversely affect our operating results.
Certain of our officers may have a conflict of interest.
Certain of our officers are currently working for our company on a part-time basis and we expect that they will continue to do so. Our employment agreement with our Chief Financial Officer/Chief Operating Officer provides that he will devote 75% of his business time to our matters, with his remaining business time devoted to other matters including, without limitation, employment at other companies that are non-competitive with us, which may result in a lack of availability when needed due to responsibilities with other requirements. Our consulting agreement with our Chief Medical Officer provides that he will devote 75% of his business time to our matters, with his remaining business time devoted to other matters including, without limitation, employment at other companies that are non-competitive with us, which may result in a lack of availability when needed due to responsibilities with other requirements.
We may acquire other businesses or form joint ventures or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, increase our debt or cause us to incur significant expense.
As part of our business strategy, we may pursue acquisitions of businesses and assets, such as the Acquisition of Purnovate. We also may pursue strategic alliances and joint ventures that leverage our technology and industry experience to expand our offerings or other capabilities. Though certain company personnel have business development and corporate transaction experience, including with licensing, mergers and acquisitions, and strategic partnering, as a company we have no experience with acquiring other companies and limited experience with forming strategic alliances and joint ventures. We may not be able to find suitable partners or acquisition candidates, and we may not be able to complete such transactions on favorable terms, if at all. If we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. Any future acquisitions also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could have a material adverse effect on our financial condition, results of operations and cash flows. Integration of an acquired company also may disrupt ongoing operations and require management resources that would otherwise focus on developing our existing business. We may experience losses related to investments in other companies, which could have a material negative effect on our results of operations. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, technology license, strategic alliance or joint venture.
To finance any acquisitions or joint ventures, we may choose to issue shares of our common stock as consideration, which would dilute the ownership of our stockholders. If the price of our common stock is low or volatile, we may not be able to acquire other companies or fund a joint venture project using our stock as consideration. Alternatively, it may be necessary for us to raise additional funds for acquisitions through public or private financings. Additional funds may not be available on terms that are favorable to us, or at all.
Declining general economic or business conditions may have a negative impact on our business.
Continuing concerns over U.S. health care reform legislation and energy costs, geopolitical issues, the availability and cost of credit and government stimulus programs in the United States and other countries have contributed to increased volatility and diminished expectations for the global economy. These factors, combined with low business and consumer confidence and high unemployment, precipitated an economic slowdown and recession and stagnant economy for more than a decade. Additionally, political changes in the U.S. and elsewhere in the world have created a level of uncertainty in the markets. If the economic climate does not improve or deteriorate, our business, as well as the financial condition of our suppliers and our third-party payors, could be adversely affected, resulting in a negative impact on our business, financial condition and results of operations.
Health care policy changes, including legislation reforming the U.S. health care system and other legislative initiatives, may have a material adverse effect on our financial condition, results of operations and cash flows.
Government payors, such as Medicare and Medicaid, have taken steps and can be expected to continue to take steps to control the cost, utilization and delivery of health care services, including clinical laboratory test services.
In March 2010, U.S. President Barack Obama signed the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the ACA, which made a number of substantial changes in the way health care is financed by both governmental and private insurers. It is unclear what, if any, changes the new administration will make to the health care system. We cannot predict whether future health care initiatives will be implemented at the federal or state level, or how any future legislation or regulation may affect us.
Risks Related to Our Securities and Investing in Our Securities
Certain of our shareholders have sufficient voting power to make corporate governance decisions that could have a significant influence on us and the other stockholders.
Our officers and directors currently beneficially own (would own, if they collectively exercised all owned warrants and options exercisable within 60 days) approximately 29% of our outstanding common stock. Bankole Johnson, our Chief Medical Officer and our former Chairman of the Board of Directors, Mr. Stilley, our Chief Executive Officer and a director, Kevin Schuyler, a director, and James W. Newman, a director, beneficially own approximately 7.2%, 10.1%, 8.1%, and 4.3%, respectively, of our common stock. As a result, our directors currently have significant influence over our management and affairs and over matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. In addition, this concentration of ownership may delay or prevent a change in our control and might affect the market price of our common stock, even when a change in control may be in the best interest of all stockholders. Furthermore, the interests of this concentration of ownership may not always coincide with our interests or the interests of other stockholders. Accordingly, these stockholders could cause us to enter into transactions or agreements that we would not otherwise consider.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans and outstanding warrants could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital may be needed in the future to continue our planned operations, including conducting clinical trials, commercialization efforts, expanded research and development activities and costs associated with operating a public company. To raise capital, we may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights, preferences and privileges senior to the holders of our common stock. Pursuant to our 2017 equity incentive plan, which became effective on the business day prior to the public trading date of our common stock, our management is authorized to grant equity awards to our employees, officers, directors and consultants.
Initially, the aggregate number of shares of our common stock that might be issued pursuant to stock awards under our 2017 equity incentive plan was 1,750,000 shares, which has been since increased to 5,500,000 at our 2020 Annual Stockholders Meeting, and of which 1,028,382 remain available for grant as of the date hereof. Increases in the number of shares available for future grant or purchase may result in additional dilution, which could cause our stock price to decline.
At December 31, 2020, we had outstanding (i) warrants to purchase 8,649,625 shares of common stock outstanding at exercise prices ranging from $0.005 to $7.634 (with a weighted average exercise price of $4.60), and (ii) options to purchase 2,668,866 shares of common stock at a weighted average exercise price of $2.48 per share. The issuance of the shares of common stock underlying the options and warrants will have a dilutive effect on the percentage ownership held by holders of our common stock.
At the date of this prospectus, having issued a significant number of options and seen a significant number of warrant exercised, we had outstanding (i) warrants to purchase 7,957,225 shares of common stock outstanding at exercise prices ranging from $0.005 to $7.634 (with a weighted average exercise price of $4.83), and (ii) options to purchase 3,576,866 shares of common stock at a weighted average exercise price of $2.61 per share. The issuance of the shares of common stock underlying the options and warrants will have a dilutive effect on the percentage ownership held by holders of our common stock.
We have additional securities available for issuance, which, if issued, could adversely affect the rights of the holders of our common stock.
Our Certificate of Incorporation authorizes the issuance of 50,000,000 shares of common stock and 5,000,000 shares of preferred stock. The common stock and preferred stock, as well as the awards available for issuance under our 2017 equity incentive plan, can be issued by our board of directors, without stockholder approval. Any future issuances of such stock would further dilute the percentage ownership in us held by holders of our common stock and may be issued at prices below the initial price offering. In addition, the issuance of preferred stock may be used as an “anti-takeover” device without further action on the part of our stockholders, and may adversely affect the holders of the common stock.
If we issue preferred stock with superior rights than our common stock, it could result in a decrease in the value of our common stock and delay or prevent a change in control of us.
Our board of directors is authorized to issue 5,000,000 shares of preferred stock in series. The issuance of any preferred stock having rights superior to those of the common stock may result in a decrease in the value or market price of our common stock. Holders of preferred stock may have the right to receive dividends, certain preferences in liquidation and conversion rights and rights to elect directors. The issuance of preferred stock could, under certain circumstances, have the effect of delaying, deferring or preventing a change in control of us without further vote or action by the stockholders and may adversely affect the voting and other rights of the holders of our common stock.
We have never paid dividends and have no plans to pay dividends in the future.
Holders of our common stock are entitled to receive such dividends as may be declared by our board of directors. To date, we have paid no cash dividends on our preferred or common stock and we do not expect to pay cash dividends in the foreseeable future. We intend to retain future earnings, if any, to provide funds for operations of our business. Therefore, any return investors in our preferred or common stock may have will be in the form of appreciation, if any, in the market value of their common stock.
Our failure to meet the continued listing requirements of The Nasdaq Capital Market could result in a de-listing of our common stock.
Our shares of common stock are listed for trading on The Nasdaq Capital Market under the symbol “ADIL” and our warrants issued in connection with our initial public offering are listed for trading on The Nasdaq Capital Market under the symbol “ADILW.” If we fail to satisfy the continued listing requirements of The Nasdaq Capital Market such as the corporate governance requirements, the stockholder’s equity requirement or the minimum closing bid price requirement, The Nasdaq Capital Market may take steps to de-list our common stock or warrants. Such a de-listing or even notification of failure to comply with such requirements would likely have a negative effect on the price of our common stock and warrants would impair your ability to sell or purchase our common stock when you wish to do so. In the event of a de-listing, we would take actions to restore our compliance with The Nasdaq Capital Market’s listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below The Nasdaq Capital Market, minimum bid price requirement or prevent future non-compliance with The Nasdaq Capital Market’s listing requirements.
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Because our common stock is listed on The Nasdaq Capital Market, our common stock is covered securities. Although the states are preempted from regulating the sale of covered securities, the federal statute does allow the states to investigate companies if there is a suspicion of fraud, and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. Further, if we were to be delisted from The Nasdaq Capital Market, our common stock would cease to be recognized as covered securities and we would be subject to regulation in each state in which we offer our securities.
We are an “emerging growth company,” and we cannot be certain if the reduced SEC reporting requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an “emerging growth company” as defined in the JOBS Act. We will remain an “emerging growth company” until the earliest to occur of (i) the last day of the fiscal year during which we have total annual gross revenue of $1.07 billion or more (subject to adjustment for inflation), (ii) the last day of the fiscal year following the fifth anniversary of the first sale of our common stock pursuant to an effective registration statement, (iii) the date on which we have, during the previous 3-year period, issued more than $1.0 billion in non-convertible debt, or (iv) the date on which we are deemed to be a “large accelerated filer.” We intend to take advantage of exemptions from various reporting requirements that are applicable to most other public companies, whether or not they are classified as “emerging growth companies,” including, but not limited to, an exemption from the provisions of Section 404(b) of Sarbanes-Oxley requiring that our independent registered public accounting firm provide an attestation report on the effectiveness of our internal control over financial reporting. An attestation report by our auditor would require additional procedures by them that could detect problems with our internal control over financial reporting that are not detected by management. If our system of internal control over financial reporting is not determined to be appropriately designed or operating effectively, it could require us to restate financial statements, cause us to fail to meet reporting obligations, and cause investors to lose confidence in our reported financial information. The JOBS Act also provides that an “emerging growth company” can take advantage of the extended transition period provided in the Securities Act, for complying with new or revised accounting standards. However, we have chosen to “opt out” of this extended transition period and, as a result, we will comply with new or revised accounting standards on or prior to the relevant dates on which adoption of such standards is required for all public companies that are not emerging growth companies. Our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable. We cannot predict if investors will find our common stock less attractive because we intend to rely on certain of these exemptions and benefits under the JOBS Act.
As a result of being a public company, we are subject to additional reporting and corporate governance requirements that will require additional management time, resources and expense.
As a public company, and particularly after we are no longer an emerging growth company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. The Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of The Nasdaq Capital Market and other applicable securities rules and regulations impose various requirements on public companies, including the obligation to file with the SEC annual and quarterly information and other reports that are specified in the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and to establish and maintain effective disclosure and financial controls and corporate governance practices. Our management and other personnel need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations increase our legal and financial compliance costs and will make some activities more time-consuming and costly.
We cannot predict or estimate the amount of additional costs we may incur or the timing of such costs. These rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
Our common stock has often been thinly traded, so you may be unable to sell at or near ask prices or at all if you need to sell your shares to raise money or otherwise desire to liquidate your shares.
To date, there have been many days on which limited trading of our common stock took place. We cannot predict the extent to which investors’ interests will lead to an active trading market for our common stock or whether the market price of our common stock will be volatile. If an active trading market does not develop, investors may have difficulty selling any of our common stock that they buy. We are likely to be too small to attract the interest of many brokerage firms and analysts. We cannot give you any assurance that an active public trading market for our common stock will develop or be sustained. The market price of our common stock could be subject to wide fluctuations in response to quarterly variations in our revenues and operating expenses, announcements of new products or services by us, significant sales of our common stock, including “short” sales, the operating and stock price performance of other companies that investors may deem comparable to us, and news reports relating to trends in our markets or general economic conditions.
Our stock price has fluctuated in the past, has recently been volatile and may be volatile in the future, and as a result, investors in our common stock could incur substantial losses.
The trading price of our common stock has been and is expected to continue to be volatile and has been and may continue to be subject to wide fluctuations in response to various factors, some of which are beyond our control, including limited trading volume. On February 16, 2021, the reported low sale price of our common stock was $2.88, the reported high sale price was $3.30 and closing price of our common stock was $3.19 while on December 31, 2020 the closing price of our common stock was $1.70. We may incur rapid and substantial decreases in our stock price in the foreseeable future that are unrelated to our operating performance for prospects. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this prospectus and the documents incorporated by reference into this prospectus, these factors include:
| | ● | the commencement, enrollment or results of the planned clinical trials of AD04 or any future clinical trials we may conduct, or changes in the development status of AD04 or any product candidates; |
| | ● | any delay in our regulatory filings for our product candidate and any adverse development or perceived adverse development with respect to the applicable regulatory authority’s review of such filings, including without limitation the FDA’s issuance of a “refusal to file” letter or a request for additional information; |
| | ● | adverse results or delays in clinical trials; |
| | ● | our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial; |
| | ● | adverse regulatory decisions, including failure to receive regulatory approval of our product candidate; |
| | ● | changes in laws or regulations applicable to our products, including but not limited to clinical trial requirements for approvals; |
| | ● | adverse developments concerning our manufacturers; |
| | ● | our inability to obtain adequate product supply for any approved product or inability to do so at acceptable prices; |
| | ● | our inability to establish collaborations if needed; |
| | ● | our failure to commercialize AD04; |
| | ● | additions or departures of key scientific or management personnel; |
| | ● | unanticipated serious safety concerns related to the use of AD04; |
| | ● | introduction of new products or services offered by us or our competitors; |
| | ● | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
| | ● | our ability to effectively manage our growth; |
| | ● | the size and growth of our initial target markets; |
| | ● | our ability to successfully treat additional types of indications or at different stages; |
| | ● | actual or anticipated variations in quarterly operating results; |
| | ● | our failure to meet the estimates and projections of the investment community or that we may otherwise provide to the public; |
| | ● | publication of research reports about us or our industry, or positive or negative recommendations or withdrawal of research coverage by securities analysts; |
| | ● | changes in the market valuations of similar companies; |
| | ● | overall performance of the equity markets; |
| | ● | sales of our common stock by us or our stockholders in the future; |
| | ● | trading volume of our common stock and declines in the market prices of stocks generally; |
| | ● | changes in accounting practices; |
| | ● | ineffectiveness of our internal controls; |
| | ● | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our or our licensee’s technologies; |
| | | |
| | ● | significant lawsuits, including patent or stockholder litigation; |
| | ● | general political and economic conditions; and |
| | ● | other events or factors, many of which are beyond our control, including those resulting from such events, or the prospect of such events, including war, terrorism and other international conflicts, public health issues including health epidemics or pandemics, such as the recent outbreak of the novel coronavirus (COVID-19), and natural disasters such as fire, hurricanes, earthquakes, tornados or other adverse weather and climate conditions, whether occurring in the United States or elsewhere, could disrupt our operations, disrupt the operations of our suppliers or result in political or economic instability. |
In addition, the stock market in general, and The Nasdaq Capital Market and biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. Since the stock price of our common stock has fluctuated in the past, has recently been volatile and may be volatile in the future, investors in our common stock could incur substantial losses. In the past, securities class action litigation has often been instituted against companies following periods of volatility in the market price of a company’s securities. This type of litigation, if instituted, could result in substantial costs and a diversion of management’s attention and resources, which would harm our business, operating results or financial condition.
Our need for future financing may result in the issuance of additional securities which will cause investors to experience dilution.
Our cash requirements may vary from those now planned depending upon numerous factors, including the result of future research and development activities. We will require additional funds in the future to complete our clinical trials of AD04. There are no other commitments by any person for future financing. Though we believe a successful Phase 3 trial will be a significant value creation event for us, our securities may be offered to other investors at a price lower than the price per share on The Nasdaq Capital Market, or upon terms which may be deemed more favorable than offered previously. In addition, the issuance of securities in any future financing using our securities may dilute an investor’s equity ownership. Moreover, we may issue derivative securities, including options and/or warrants, from time to time, to procure qualified personnel or for other business reasons. The issuance of any such derivative securities, which is at the discretion of our board of directors, may further dilute the equity ownership of our stockholders. No assurance can be given as to our ability to procure additional financing, if required, and on terms deemed favorable to us. To the extent additional capital is required and cannot be raised successfully, we may then have to limit our then current operations and/or may have to curtail certain, if not all, of our business objectives and plans.
Fluctuations in the international currency markets may significantly impact the cost of our planned Phase 3 trial.
Many of the costs associated with our ongoing ONWARD Phase 3 trial, presently expected to require approximately $10.7 million to complete, are denominated in Euros, while our funding is held in US Dollars. A change in the value of the Euro relative to the US Dollar may significantly impact the cost of our trial, positively or negatively.
The application of the “penny stock” rules to our common stock could limit the trading and liquidity of the common stock, adversely affect the market price of our common stock and increase your transaction costs to sell those shares.
If our common stock is no longer listed on The Nasdaq Capital Market and becomes traded on a securities market or exchange which is not registered as a national securities exchange with the SEC under Section 6 of the Exchange Act, as long as the trading price of our common stock is below $5 per share, the open-market trading of our common stock will be subject to the “penny stock” rules, unless we otherwise qualify for an exemption from the “penny stock” definition. The “penny stock” rules impose additional sales practice requirements on certain broker-dealers who sell securities to persons other than established customers and accredited investors (generally those with assets in excess of $1.0 million or annual income exceeding $200,000 or $300,000 together with their spouse). These regulations, if they apply, require the delivery, prior to any transaction involving a penny stock, of a disclosure schedule explaining the penny stock market and the associated risks. Under these regulations, certain brokers who recommend such securities to persons other than established customers or certain accredited investors must make a special written suitability determination regarding such a purchaser and receive such purchaser’s written agreement to a transaction prior to sale. These regulations may have the effect of limiting the trading activity of our common stock, reducing the liquidity of an investment in our common stock and increasing the transaction costs for sales and purchases of our common stock as compared to other securities. The stock market in general and the market prices for penny stock companies in particular, have experienced volatility that often has been unrelated to the operating performance of such companies. These broad market and industry fluctuations may adversely affect the price of our stock, regardless of our operating performance. Stockholders should be aware that, according to SEC Release No. 34-29093, the market for penny stocks has suffered in recent years from patterns of fraud and abuse. Such patterns include: (i) control of the market for the security by one or a few broker-dealers that are often related to the promoter or issuer; (ii) manipulation of prices through prearranged matching of purchases and sales and false and misleading press releases; (iii) boiler room practices involving high-pressure sales tactics and unrealistic price projections by inexperienced sales persons; (iv) excessive and undisclosed bid-ask differential and markups by selling broker-dealers; and (v) the wholesale dumping of the same securities by promoters and broker-dealers after prices have been manipulated to a desired level, along with the resulting inevitable collapse of those prices and with consequent investor losses. The occurrence of these patterns or practices could increase the volatility of our share price.
Provisions in our corporate charter documents and under Delaware law could make an acquisition of our company, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our corporate charter and our bylaws may discourage, delay or prevent a merger, acquisition or other change in control of our company that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, because our board of directors is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Among other things, these provisions:
| | ● | our board of directors is divided into three classes, one class of which is elected each year by our stockholders with the directors in each class to serve for a three-year term; |
| | ● | the authorized number of directors can be changed only by resolution of our board of directors; |
| | ● | directors may be removed only by the affirmative vote of the holders of at least sixty percent (60%) of our voting stock, whether for cause or without cause; |
| | | |
| | ● | our bylaws may be amended or repealed by our board of directors or by the affirmative vote of sixty-six and two-thirds percent (66 2/3%) of our stockholders; |
| | | |
| | ● | stockholders may not call special meetings of the stockholders or fill vacancies on the board of directors; |
| | ● | our board of directors will be authorized to issue, without stockholder approval, preferred stock, the rights of which will be determined at the discretion of the board of directors and that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that our board of directors does not approve; |
| | ● | our stockholders do not have cumulative voting rights, and therefore our stockholders holding a majority of the shares of common stock outstanding will be able to elect all of our directors; and |
| | ● | our stockholders must comply with advance notice provisions to bring business before or nominate directors for election at a stockholder meeting. |
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Our Certificate of Incorporation and our bylaws provide that the Court of Chancery of the State of Delaware will be the exclusive forum for certain types of state actions that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, or employees.
Our Certificate of Incorporation and our bylaws provide that, unless we consent to the selection of an alternative forum, the Court of Chancery of the State of Delaware is the exclusive forum for (i) any derivative action or proceeding brought on behalf of us, (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers, or other employees to us or our stockholders, (iii) any action arising pursuant to any provision of the DGCL or our certificate of incorporation or bylaws (as either may be amended from time to time), or (iv) any action asserting a claim governed by the internal affairs doctrine. The exclusive forum provision does not apply to suits brought to enforce any liability or duty created by the Securities Act or the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. To the extent that any such claims may be based upon federal law claims, Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. Furthermore, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder.
These exclusive-forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, employees, control persons, underwriters, or agents, which may discourage lawsuits against us and our directors, employees, control persons, underwriters, or agents. Additionally, a court could determine that the exclusive forum provision is unenforceable, and our stockholders will not be deemed to have waived our compliance with the federal securities laws and the rules and regulations thereunder. If a court were to find these provisions of our bylaws inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business, financial condition, or results of operations.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on our company. If no securities or industry analysts commence coverage of our company, the trading price for our stock would likely be negatively impacted. In the event securities or industry analysts initiate coverage, if one or more of the analysts who covers us downgrades our stock or publishes inaccurate or unfavorable research about our business, our stock price may decline. If one or more of these analysts ceases coverage of our company or fails to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and trading volume to decline.
The warrants that we have issued are speculative in nature.
The warrants that we have issued do not confer any rights of common stock ownership on their holders except as otherwise provided in the warrants. Specifically, commencing on the date of issuance, holders of the warrants may exercise their right to acquire the common stock and pay the exercise price to acquire the warrants. There can be no assurance that the market value of the warrants will equal or exceed their public offering price. In the event our common stock price does not exceed the exercise price of the warrants during the period when the warrants are exercisable, the warrants may not have any value.
Holders of the warrants will have no rights as a common stockholder except as otherwise provided in the warrants until they acquire our common stock.
Until holders of warrants acquire shares of our common stock upon exercise of their warrants, they will have no rights with respect to shares of our common stock issuable upon exercise of their warrant except as otherwise provided in the warrant. Upon exercise of a warrant, a holder will be entitled to exercise the rights of a common stockholder as to the security exercised only as to matters for which the record date occurs after the exercise.
There is no established market for the warrants issued in our follow-on offering and those issued prior to our initial public offering.
There is no established trading market for the warrants issued in our follow-on offering and those issued prior to our initial public offering and we do not expect a market to develop. We have not applied for the listing of such warrants on any national securities exchange or other trading market. Without an active trading market, the liquidity of the warrants will be limited.
Provisions of the warrants issued in our public offerings could discourage an acquisition of us by a third party.
In addition to the discussion of the provisions of our certificate of incorporation, our bylaws, certain provisions of the warrants offered in our public offerings could make it more difficult or expensive for a third party to acquire us. The warrants prohibit us from engaging in certain transactions constituting “fundamental transactions” unless, among other things, the surviving entity assumes our obligations under the warrants. These and other provisions of the warrants could prevent or deter a third party from acquiring us even where the acquisition could be beneficial to you.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference herein contain forward-looking statements that are based on current management expectations. Statements other than statements of historical fact included in this prospectus, including statements about us and the future growth and anticipated operating results and cash expenditures, are forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Exchange Act. When used in this prospectus the words “anticipate,” “objective,” “may,” “might,” “should,” “could,” “can,” “intend,” “expect,” “believe,” “estimate,” “predict,” “potential,” “plan” or the negative of these and similar expressions identify forward-looking statements. These statements reflect our current views with respect to uncertain future events and are based on imprecise estimates and assumptions and subject to risk and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. While we believe our plans, intentions and expectations reflected in those forward-looking statements are reasonable, these plans, intentions or expectations may not be achieved. Our actual results, performance or achievements could differ materially from those contemplated, expressed or implied by the forward-looking statements contained in, or incorporated by reference into, this prospectus for a variety of reasons. Those risks and uncertainties include, among others:
| | ● | our ability to implement our business plan; |
| | | |
| | ● | our ability to raise additional capital to meet our liquidity needs; |
| | | |
| | ● | our ability to generate product revenues; |
| | | |
| | ● | our ability to achieve profitability; |
| | | |
| | ● | our ability to satisfy U.S. (including FDA) and international regulatory requirements; |
| | | |
| | ● | our ability to obtain market acceptance of our technology and products; |
| | | |
| | ● | our ability to compete in the market; |
| | | |
| | ● | our ability to advance our clinical trials; |
| | | |
| | ● | our ability to fund, design and implement clinical trials; |
| | | |
| | ● | our ability to demonstrate that our lead product candidate is safe for human use and effective for indicated uses; |
| | | |
| | ● | our ability to gain acceptance of physicians and patients for use of our lead product; |
| | | |
| | ● | our dependency on third-party researchers, manufacturers and payors; |
| | | |
| | ● | our ability to establish and maintain strategic partnerships, including for the distribution of our lead product and any future products that we may acquire; |
| | | |
| | ● | our ability to attract and retain a sufficient qualified personnel; |
| | | |
| | ● | our ability our ability to obtain or maintain patents or other appropriate protection for the intellectual property; |
| | | |
| | ● | our dependency on the intellectual property licensed to us or possessed by third parties; |
| | | |
| | ● | our ability to adequately support future growth; |
| | | |
| | ● | potential product liability or intellectual property infringement claims; |
| | | |
| | ● | our ability to successfully integrate the Purnovate business into our business; and |
| | | |
| | ● | disruption or delay of our ongoing clinical trial, disruption of our corporate operations or those of our critical vendors, or general significant disruption to the global economy as a result of the outbreak of the novel coronavirus (COVID-19) pandemic. |
We urge investors to review carefully risks contained in the section of this prospectus entitled “Risk Factors” above as well as other risks and factors identified from time to time in our SEC filings in evaluating the forward-looking statements contained in this prospectus. We caution investors not to place significant reliance on forward-looking statements contained in this document; such statements need to be evaluated in light of all the information contained herein.
All forward-looking statements attributable to us or persons acting on our behalf are expressly qualified in their entirety by the risk factors and other cautionary statements set forth, or incorporated by reference, in this prospectus. Other than as required by applicable securities laws, we are under no obligation, and we do not intend, to update any forward-looking statement, whether as result of new information, future events or otherwise.
USE OF PROCEEDS
We are not selling any shares of our common stock in this offering and, as a result, we will not receive any proceeds from the sale of the common stock covered by this prospectus. All of the net proceeds from the sale of our common stock will go to the warrant holders. Upon exercise of the warrants, however, we will receive proceeds from the exercise of such warrants if exercised for cash and not on a cashless basis. We will receive approximately $9,844,450 of proceeds if all the currently outstanding warrants issued in the February 2019 public offering are exercised for cash. We currently intend to use these proceeds for general corporate purposes.
DIVIDEND POLICY
We do not anticipate paying dividends on our common stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. We are not subject to any legal restrictions respecting the payment of dividends, except that we may not pay dividends if the payment would render us insolvent. Any future determination as to the payment of cash dividends on our common stock will be at our board of directors’ discretion and will depend on our financial condition, operating results, capital requirements and other factors that our board of directors considers to be relevant.
MARKET FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
Market Information
On July 27, 2018, our common stock and our warrants issued in connection with our July 2018 initial public offering began trading on The Nasdaq Capital Market under the symbols “ADIL” and “ADILW,” respectively. Prior to our initial public offering, no public trades occurred in our common stock or warrants. On March 22, 2021, the last reported sale price of our common stock on the Nasdaq Capital Market was $2.51 per share. The last reported sale price of our warrants issued in the initial public offering on March 22, 2021 was $0.6301 per warrant. We urge prospective purchasers of our common stock to obtain current information about the market prices of our common stock and our warrants issued in connection with our initial public offering in July 2018.
Dividend Policy
We have not paid dividends on our common stock to date and do not anticipate paying dividends on our common stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. We are not subject to any legal restrictions respecting the payment of dividends, except that we may not pay dividends if the payment would render us insolvent. Any future determination as to the payment of cash dividends on our common stock will be at our board of directors’ discretion and will depend on our financial condition, operating results, capital requirements and other factors that our board of directors considers to be relevant.
Transfer Agent, Warrant Agent and Registrar
The transfer agent and registrar for our common stock and warrant agent for our warrants offered in our initial public offering is VStock Transfer, LLC.
Holders of Common Stock and Warrants
As of March 22, 2021, there were an estimated 77 holders of record of our common stock and 38 holders of record of our warrants issued in connection with our initial public offering. A certain amount of the shares of common stock are held in street name and may, therefore, be held by additional beneficial owners. This number does not include beneficial owners from whom shares are held by nominees in street name.
Performance Graph and Purchases of Equity Securities
The Company is a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and is not required to provide the information required under this item.
DILUTION
If you invest in our securities, your interest may be immediately and substantially diluted to the extent of the difference between the price you pay per share and the pro forma net tangible book value per share of our common stock.
Our historical net tangible book value as December 31, 2020 was $3,245,736, or $0.23 per share.
Our pro forma net tangible book value as of December 31, 2020 was $6,976,239, or $0.40 per share and includes the following transactions subsequent to December 31, 2020: (i) gross proceeds of approximately $2,350,000 from the issuance and sale of 1,007,296 shares of our common stock pursuant to our equity line with Keystone Capital, (ii) proceeds of $1,425,000 from the exercise of warrants to purchase 712,500 shares of our common stock for an exercise price of $2.00 per share, (iii) gross proceeds of $291,003 from the issuance of 97,001 shares of our common stock in a private placement pursuant to the Securities Purchase Agreements, (iv) the issuance of approximately 700,000 shares of our common stock and the payment of $350,000 upon the closing of the Purnovate acquisition, (v) proceeds of $14,500 from the exercise of 10,000 options to purchase shares of common stock at $1.45 per share, and (vi) issuance of a total of 350,000 shares of common stock to consultants and vendors for services provided.
Assuming the exercise of all of the remaining outstanding warrants issued in the initial public offering, which means the issuance of 1,575,112 shares of common stock upon the exercise of such warrants, our pro forma as adjusted net tangible book value would have been approximately $16,820,689, or $0.89 per share at December 31, 2020. This represents an immediate increase in net tangible book value of approximately $0.49 per share to our existing stockholders, and an immediate dilution of $5.36 per share to investors purchasing shares in an offering using this prospectus, assuming an exercise price per share of $6.25 per share.
Dilution in pro forma as adjusted net tangible book value per share represents the difference between the amount per share of common stock and the pro forma net tangible book value per share of our common stock immediately after this offering.
The following table illustrates the per share dilution to investors purchasing shares in an offering using this prospectus:
| Assumed price per share | | | | | | $ | 6.25 | |
| Pro forma net tangible book value per share as of December 31, 2020 | | $ | 0.40 | | | | | |
| Increase in pro forma net tangible book value per share | | $ | 0.49 | | | | | |
| Pro forma as adjusted net tangible book value per share after this offering | | | | | | $ | 0.89 | |
| Dilution per share to new investors | | | | | | $ | 5.36 | |
The number of shares of common stock that will be outstanding immediately after this offering is based on 14,393,100 shares of common stock outstanding as of December 31, 2020. This amount excludes, as of December 31, 2020:
| | ● | 2,171,382 shares of our common stock reserved for future issuance under our 2017 equity incentive plan; |
| | ● | 8,649,625 shares of our common stock issuable upon exercise of the warrants at a weighted average exercise price of $4.60 per share (not including warrants registered under the Prior Registration Statement); and |
| | ● | 2,668,866 shares of our common stock issuable upon outstanding options to purchase shares of common stock at a weighted average exercise price of $2.61 per share. |
Management’s Discussion and Analysis of Financial Condition and Results
of Operations
The following discussion and analysis is intended as a review of significant factors affecting our financial condition and results of operations for the periods indicated. The discussion should be read in conjunction with our financial statements and the notes presented herein. In addition to historical information, the following Management’s Discussion and Analysis of Financial Condition and Results of Operations contains forward-looking statements that involve risks and uncertainties. See “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements” included elsewhere in this registration statement. Our actual results could differ significantly from those expressed, implied or anticipated in these forward-looking statements as a result of certain factors discussed herein and any other periodic reports filed and to be filed by us with the Securities and Exchange Commission.
Overview
We are a clinical-stage biopharmaceutical company focused on the development of therapeutics for the treatment or prevention of addiction and related disorders. Our lead investigational new drug product, AD04, is being developed as a therapeutic agent for the treatment of alcohol use disorder (“AUD”). The active ingredient in AD04 is ondansetron, a selective serotonin-3 antagonist (i.e., a “5-HT3 antagonist”) that is also the active ingredient in Zofran®, an approved drug for treating nausea and emesis. We have commenced Phase 3 clinical trial using AD04 for the potential treatment of AUD in subjects with certain target genotypes. We believe our approach is unique in that it targets the serotonin system and individualizes the treatment of AUD, through the use of genetic screening (i.e., a companion diagnostic genetic biomarker). We have created an investigational companion diagnostic biomarker test for the genetic screening of patients with certain biomarkers that, as reported in the American Journal of Psychiatry (Johnson, et. al. 2011 & 2013), we believe will benefit from treatment with AD04. Our strategy is to integrate the pre-treatment genetic screening into AD04’s label to create a patient-specific treatment in one integrated therapeutic offering. Our goal is to develop a genetically targeted, effective and safe product candidate to treat AUD by reducing or eliminating the patients’ consumption of alcohol. We are also exploring expanding or portfolio in the field of addiction.
We have a worldwide, exclusive license from the University of Virginia Patent Foundation (d.b.a the Licensing & Venture Group) (“UVA LVG”), which is the licensing arm of the University of Virginia, to commercialize our investigational drug candidate, AD04, subject to Food and Drug Administration (“FDA”) approval of the product, based upon three separate patent application families, with patents issued in over 40 jurisdictions, including three issued patents in the U.S. Our investigational agent has been used in several investigator-sponsored trials and we possess or have rights to use toxicology, pharmacokinetic and other preclinical and clinical data that supports our Phase 3 clinical trial. Our therapeutic agent was the product candidate used in a University of Virginia investigator sponsored Phase 2b clinical trial of 283 patients. In this Phase 2b clinical trial, ultra-low dose ondansetron, the active pharmaceutical agent in AD04, patients with the target genotypes showed a statistically significant difference between ondansetron and placebo for both the primary endpoint and secondary endpoint, which were reduction in severity of drinking measured in drinks per drinking day (1.71 drinks/drinking day; p=0.0042), and reduction in frequency of drinking measured in days of abstinence/no drinking (11.56%; p=0.0352), respectively. Additionally, and importantly, the Phase 2b results showed a significant decrease in the percentage of heavy drinking days (11.08%; p=0.0445) with a “heavy drinking day” defined as a day with four (4) or more alcoholic drinks for women or five (5) or more alcoholic drinks for men consumed in the same day.
The active pharmaceutical agent in AD04, our lead investigational new drug product, is ondansetron (the active ingredient in Zofran®), which was granted FDA approval in 1991 for nausea and vomiting post-operatively and after chemotherapy or radiation treatment and is now commercially available in generic form. In studies of Zofran®, conducted as part of its FDA review process, ondansetron was given acutely at dosages up to almost 100 times the dosage expected to be formulated in AD04 with the highest doses of Zofran® given intravenously (“i.v.”), which results in approximately 160% of the exposure level as oral dosing. Even at high doses given i.v. the studies found that ondansetron is well-tolerated and results in few adverse side effects at the currently marketed doses, which reach more than 80 times the AD04 dose and are given i.v. The formulation dosage of ondansetron used in our drug candidate (and expected to be used by us in our Phase 3 clinical trials) has the potential advantage that it contains a much lower concentration of ondansetron than the generic formulation/dosage that has been used in prior clinical trials, is dosed orally, and is available with use of a companion diagnostic genetic biomarker. Our development plan for AD04 is designed to demonstrate both the efficacy of AD04 in the genetically targeted population and the safety of ondansetron when administered chronically at the AD04 dosage. However, to the best of our knowledge, no comprehensive clinical study has been performed to date that has evaluated the safety profile of ondansetron at any dosage for long-term use as anticipated in our Phase 3 clinical trial.
According to the National Institute of Alcohol Abuse and Alcoholism (the “NIAAA”) and the Journal of the American Medical Association (“JAMA”), in the United States alone, approximately 35 million people each year have AUD (such number is based upon the 2012 data provided in Grant et. al. the JAMA 2015 publication and has been adjusted to reflect a compound annual growth rate of 1.13%, which is the growth rate reported by U.S. Census Bureau for the general adult population from 2012-2017), resulting in significant health, social and financial costs with excessive alcohol use being the third leading cause of preventable death and is responsible for 31% of driving fatalities in the United States (NIAAA Alcohol Facts & Statistics). AUD contributes to over 200 different diseases and 10% of children live with a person that has an alcohol problem. According to the American Society of Clinical Oncologists, 5-6% of new cancers and cancer deaths globally are directly attributable to alcohol. And, The Lancet published that alcohol is the leading cause of death in people ages 15-49 globally. The Centers for Disease Control (the “CDC”) has reported that AUD costs the U.S. economy about $250 billion annually, with heavy drinking accounting for greater than 75% of the social and health related costs. Despite this, according to the article in the JAMA 2015 publication, only 7.7% of patients (i.e., approximately 2.7 million people) with AUD are estimated to have been treated in any way and only 3.6% by a physician (i.e., approximately 1.3 million people). In addition, according to the JAMA 2017 publication, the problem in the United States appears to be growing with almost a 50% increase in AUD prevalence between 2002 and 2013.
We have devoted substantially all of our resources to development efforts relating to AD04, including preparation for conducting clinical trials, providing general and administrative support for these operations and protecting our intellectual property. We currently do not have any products approved for sale and we have not generated any significant revenue since our inception. From our inception through the date of this Annual Report on Form 10-K, we have funded our operations primarily through the private placement of debt and equity securities and most recently, our initial public offering, follow-on offering and equity line.
We have incurred net losses in each year since our inception, including net losses of approximately $10.9 million and $8.6 million for the years ended December 31, 2020 and 2019. We had accumulated deficits of approximately $31.5 million as of December 31, 2020. Substantially all our operating losses resulted from costs incurred in connection with our research and development programs, from general and administrative costs associated with our operations, and from financing costs.
We will not generate revenue from product sales unless and until we successfully complete development and obtain marketing approval for AD04, which we expect will take a number of years and is subject to significant uncertainty. We do not believe our current cash and equivalents will be sufficient to fund our operations for the next twelve months from the filing of these financial statements, because we have incurred various expenses related to adding personnel and other corporate resources and experienced delays in certain countries in obtaining regulatory approval required to commence the trial in such countries due to COVID-19, resulting in significantly slowed trial enrollment and additional expense. We expect that we will need additional funding to complete our first Phase 3 clinical trial.
Until such time, if ever, as we can generate substantial revenue from product sales, we expect to finance our operating activities through a combination of equity offerings, debt financings, government or other third-party funding, commercialization, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements. However, we may be unable to raise additional funds or enter into such other arrangements when needed on favorable terms or at all. Our failure to raise capital or enter into such other arrangements as and when needed would have a negative impact on our financial condition and our ability to develop AD04.
Clinical Trials — Research and Development Schedule
We currently anticipate that we, working in collaboration with our vendors, upon execution of collaborative research and development agreements with them, will be able to execute the following timeline:
AD04 — Two-Stage Clinical Development Strategy — Conduct the Phase 3 clinical trials sequentially
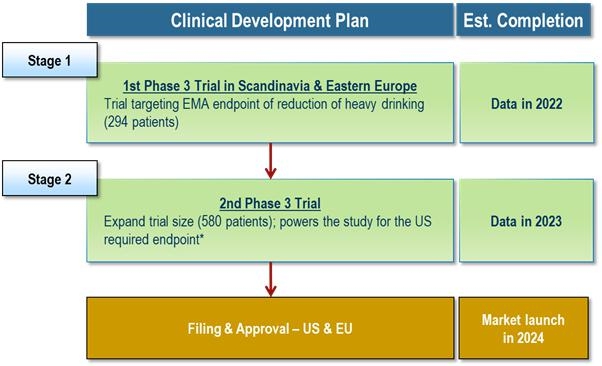
| * | Even if the 1st Phase 3 trial is not accepted by the FDA due to the study not being well-powered for the FDA’s currently stated end point, we still expect that the EMA will require only one additional trial. In this case, however, a 3rd trial might be required by the FDA (i.e., three Phase 3 trials in total). If two additional trials are required for FDA approval after an initial Phase 3 trial conducted in the EMA, we would expect to run the 2nd and 3rd trials in parallel (i.e., at the same time) so as not to increase the expected time to approval. The 2nd Phase 3 trial is expected to require $20 million in direct expenses, and up to $10 million in additional other development expenses is expected to be required. A possible 3rd Phase 3 trial would be expected to require an additional $20 million in clinical trial related expenditures. |
We current estimate the total cost to complete our initial Phase 3 clinical trial of AD04 for the treatment of AUD to be approximately $10.7 million (versus a previous estimate of $8.8 million), of which approximately $5.3 million has already been incurred or been pre-paid, leaving approximately $5.4 million in direct trial expenses that we will be required to pay in the future. This estimate is subject to many factors, some of which are beyond our control. These factors include, but are not limited to, the following:
| | ● | the progress and cost of our research and development activities; |
| | ● | the number and scope of our research and development programs; |
| | ● | the progress and cost of our preclinical and clinical development activities; |
| | ● | our ability to maintain current research and development licensing arrangements and to establish new research and development and licensing arrangements; |
| | ● | our ability to achieve our milestones under licensing arrangements; |
| | ● | the costs involved in prosecuting and enforcing patent claims and other intellectual property rights; |
| | ● | the costs and timing of regulatory approvals; |
| | ● | The impact of COVID-19 on our ability to timely enroll patients in our Phase 3clinical trial and obtain regulatory approvals; and |
| | ● | changes in the value of the Euro relative to the US Dollar. |
Additional funds are expected to be raised through grants, partnerships with other pharmaceutical companies or through additional debt or equity financings, including pursuant to the terms of our equity line. We expect the second Phase 3 Trial to cost approximately $20 million, such estimate subject to the factors stated above.
As we advance our clinical programs, we are in close contact with our CROs and clinical sites and are assessing the impact of COVID-19 on our studies and current timelines and costs.
2020 Financing Developments
On June 11, 2020, we concluded a registered direct offering of 2,820,000 shares of common stock and in a concurrent private placement the sale of warrants to purchase 2,115,000 shares of common stock at an exercise price of $2.00 per share. The shares of common stock and accompanying warrants were sold directly to the buyers at a combined at-the-market price of $1.85 for a share and three quarters warrant. Gross proceeds of the offering, totaled $5,217,000, which after offering expenses, resulted in net proceeds of $4,657,215.
On September 21, 2020, we concluded a private placement with a related party of 357,143 unregistered shares of common stock and above market price of $1.40 per share. Gross proceeds of the offering, totaled $500,000, with no material expenses.
On September 29, 2020, previously registered warrants to purchase 225,000 shares at an exercise fee of $2.00 per share were exercised for a total of $450,000.
On November 18, 2020, we entered into a purchase agreement (the “Purchase Agreement”) and a registration rights agreement (the “Registration Rights Agreement”) with Keystone Capital. Pursuant to the Purchase Agreement, we have the right to sell Keystone Capital the lesser of (i) $15,000,000 in shares of our common stock and (ii) the number of shares of common stock equal to the Exchange Cap (as defined below), subject to certain limitations and conditions set forth in the Purchase Agreement, including a closing market price of Adial stock of greater than $1.00 on the business day sales under the agreement are made. The purchase price of the shares of our common stock that may be sold to Keystone Capital under the Purchase Agreement will be based on the market price of our common stock at the time of sale as computed under the Purchase Agreement. specifically, the purchase price per share of the common stock that may be sold to Keystone Capital under the Purchase Agreement is such fixed purchases equal ninety percent (90%) of the arithmetic average of the closing sale prices of our common stock during the five (5) consecutive trading-day period ending on the fixed purchase date for the fixed purchase, so long as the common stock is listed on Nasdaq or any nationally recognized successor thereto (to be appropriately adjusted for any reorganization, recapitalization, non-cash dividend, stock split or other similar transaction that occurs on or after the date of this Purchase Agreement). There is no upper limit on the price per share that Keystone Capital could be obligated to pay for the common stock under the Purchase Agreement.
Under the applicable rules of the Nasdaq Stock Market LLC (“Nasdaq”), in no event may we issue more than 2,842,198 shares of our common stock to Keystone Capital under the Purchase Agreement (including 175,000 shares of our common stock that we issued to Keystone Capital upon execution of the Purchase Agreement, the cost of which issuance was capitalized as a cost of equity), which represents 19.99% of the shares of our common stock outstanding immediately prior to the execution of the Purchase Agreement (the “Exchange Cap”), unless (i) we obtain stockholder approval to issue shares of our common stock in excess of the Exchange Cap or (ii) the average price of all applicable sales of common stock to Keystone Capital under the Purchase Agreement equals or exceeds $1.8222, which represents the lower of (i) the Nasdaq official closing price immediately preceding the execution of the Purchase Agreement and (ii) the average of the five Nasdaq official closing prices for the common stock immediately preceding the execution of the Purchase Agreement, plus an incremental amount such that the transactions contemplated by the Purchase Agreement are exempt from the Exchange Cap limitation under applicable Nasdaq rules. In any event, the Purchase Agreement specifically provides that we may not issue or sell any shares of our Common Stock under the Purchase Agreement if such issuance or sale would breach any applicable rules or regulations of the Nasdaq. The Company has also limited the aggregate number of shares of common stock reserved for issuance under the Purchase Agreement to 15,000,000 shares without subsequent approval from our board of directors.
Pursuant to the terms of the Registration Rights Agreement, we agreed to file with the SEC one or more registration statements on Form S-1 to register for resale under the Securities Act the shares of our common stock that may be issued to Keystone Capital under the Purchase Agreement, including the commitment shares that we issued to Keystone Capital. The Purchase Agreement and the Registration Rights Agreement contain customary representations, warranties, conditions and indemnification obligations of the parties. The registration statement registering such shares of common stock was declared effective on December 15, 2020. During the year ended December 31, 2020, we did not sell any shares of our common stock pursuant to the Purchase Agreement. Subsequent to the year-end we sold 1,007,296 shares of our common stock for gross proceeds of $2,350,000 pursuant to the Purchase Agreement.
On March 11, 2021, we entered into a Securities Purchase Agreement (the “Securities Purchase Agreements”) with each of Keystone Capital Partners, LLC (“Keystone”), Bespoke Growth Partners, Inc. (“Bespoke”), a company controlled by Mark Peikin, our Chief Strategy Officer and entities controlled by James W. Newman, Jr., a member of our board of directors (“Newman” and collectively with Keystone and Bespoke the “Investors,” and each an “Investor”), pursuant to which: (i) Keystone has agreed to purchase an aggregate of 333,334 shares of our common stock at a purchase price of $3.00 per share for aggregate gross proceeds of $1,000,002; (ii) Bespoke has agreed to purchase an aggregate of 336,667 shares of our common stock at a purchase price of $3.00 per share for aggregate gross proceeds of $1,010,001; and (iii) Newman has agreed to purchase an aggregate of 30,000 shares of our common stock at a purchase price of $3.00 per share for aggregate gross proceeds of $90,000. Under the terms of the Securities Purchase Agreements: (i) Keystone has purchased 33,334 shares of our common stock and paid us $100,002 and agreed to purchase an additional 300,000 shares of our common stock upon the effectiveness of a registration statement registering the shares of common stock acquired and to be acquired (the “Registration Statement”); (ii) Bespoke has purchased 33,337 shares of our common stock and paid us $100,011 and agreed to purchase an additional 300,000 shares of our common stock upon the effectiveness of the Registration Statement; and (iii) Newman has purchased 30,000 shares of our common stock and paid us $90,000. In connection with the Securities Purchase Agreements, we entered into Registration Rights Agreements, dated March 11, 2021 (“Registration Rights Agreements”), with each of the Investors pursuant to which we are obligated to file a registration statement (the “Registration Statement”) with the SEC within thirty (30) days following the date upon which we file our Annual Report on Form 10-K for the fiscal year ended December 31, 2020 with the SEC and use all commercially reasonable efforts to have the Registration Statement declared effective by the SEC within thirty (30) days after the Registration Statement is filed (or, in the event of a “full review” by the SEC, within thirty (30) days after the Registration Statement is filed).
Recent Developments
Acquisition of Purnovate, Inc.
On December 7, 2020, we closed the Acquisition contemplated by that Equity Purchase Agreement pursuant to which we purchased all of the outstanding membership interests of Purnovate from the members of Purnovate, such that after the Acquisition, Purnovate became our wholly owned subsidiary. Purnovate is a pre-clinical drug development company with a platform focused on developing drug candidates for non-opioid pain reduction and other diseases and disorders potentially targeted with adenosine analogs that are selective, potent, stable, and soluble. Prior to the acquisition, our CEO and a Director owned equity in Purnovate and the transaction was considered to be one with a related party.
Pursuant to the terms of the Equity Purchase Agreement, in exchange for the outstanding membership interests of Purnovate, we paid the members an aggregate of $350,000 (the “Cash Consideration”) at the closing and issued to the Members an aggregate of 700,000 shares of our restricted common stock (the “Stock Consideration”) with an approximate fair market value of $1,638,000 at time of issuance, which issuance was exempt from registration pursuant to Section 4(a)(2) of the Securities Act of 1933, as amended. Prior to the closing of the transaction, we loaned Purnovate $350,000 in working capital for use during the due diligence period. In addition, members will receive development milestone payments in an aggregate amount of up to $2,100,000 for each compound developed, development milestone payments in an aggregate amount of up to $20,000,000 for each compound commercialized, and royalties of 3.0% of net sales.
Clinical Developments
In January 2020, we announced that we had received favorable opinions from the Finnish Medicines Agency (FIMEA) and National Committee on Medical Research Ethics (TUKIJA) to commence our Phase 3 clinical trial to investigate AD04 as a genetically targeted therapeutic agent for the treatment of AUD.
On June 11, 2020, we announced that we had received all necessary approvals to commence our landmark Phase 3 clinical trial to investigate AD04 as a genetically targeted therapeutic agent for the treatment of AUD.
On July 17, 2020, we received notice that the European Medicines Agency (EMA) had accepted the Pediatric Investigation Plan (PIP) submitted by us for development of our lead drug candidate, AD04, for the treatment of alcohol use disorder in the pediatric population, ages 12 to 17.
On July 30, 2020, we announced that we had received approval to commence our Phase 3 clinical trial of AD04 for the treatment of AUD in Croatia. As a result, we had secured approvals to run the trial in all of the Scandinavian and Eastern European countries in which we intend to run the clinical trial.
On September 14, 2020, we filed with the FDA to take our Investigational New Drug (IND) Application for the use of our lead product candidate, AD04, as a treatment for Alcohol Use Disorder (AUD) off inactive status with the United States Food and Drug Administration (FDA) and on October 15, 2020 we announced that that the FDA had reactivated the IND.
On November 3, 2020, we providing the following highlighted updates on our Phase 3 clinical trial of AD04 for the treatment of AUD
| | ● | All 25 planned investigative sites are active. |
| | ● | 66% of expected patient screenings have been completed and the Phase 3 clinical trial more than 50% enrolled relative to planned enrollment. |
| | ● | The enrollment rate has increased for four consecutive months and the Phase 3 clinical trial is projected to be fully enrolled during Q2 2021. |
| | ● | Data readout is expected in Q4 2021 or earlier. |
| | ● | AD04 has been well tolerated with no serious adverse events having been reported |
Antibody Tests
In order to protect our patients in the ONWARD Phase 3 trial and potentially increase retention rates, we secured distribution rights to certain SARS-CoV-2 antibody tests to administer to patients in the trial and various time points. In an effort to make the antibody tests more widely available in the United States, we entered engagements with third parties for sell tests provided by Adial as their distributor. Significant resources have not been devoted to this effort and the impact on the Company is not expected to be material.
Results of operations for the years ended December 31, 2020 and 2019 (rounded to nearest thousand)
The following table sets forth the components of our statements of operations in dollars for the periods presented:
| | | For the Year Ended
December 31, | | | Change | |
| | | 2020 | | | 2019 | | | (Decrease) | |
| Research and development expenses | | $ | 5,853,000 | | | | 3,966,000 | | | | 1,887,000 | |
| General and administrative expenses | | | 5,075,000 | | | | 4,279,000 | | | | 796,000 | |
| Total Operating Expenses | | | 10,928,000 | | | | 8,245,000 | | | | 2,683,000 | |
| | | | | | | | | | | | | |
| Loss From Operations | | | (10,928,000 | ) | | | (8,245,000 | ) | | | (2,683,000 | ) |
| | | | | | | | | | | | | |
| Interest income | | | 32,000 | | | | 95,000 | | | | (63,000 | ) |
| Warrant Modification Expense | | | – | | | | (442,000 | ) | | | 442,000 | |
| Other Income | | | 3,000 | | | | – | | | | 3,000 | |
| Total other income (expenses) | | | 35,000 | | | | (347,000 | ) | | | 382,000 | |
| | | | | | | | | | | | | |
| Net Loss | | | (10,893,000 | ) | | | (8,592,000 | ) | | | (2,301,000 | ) |
Research and development (“R&D”) expenses
Research and development expenses increased by approximately $1,887,000 (48%) during the year ended December 31, 2020 as compared to the year ended December 31, 2019. This change was primarily due to a large increase in direct trial expenses (approximately $2,967,000) consisting of CRO fees and expenses, site fees and related vendor expenses with an increase in trial sites open to enrollment, partially offset by decreases in regulatory consulting fees due to decreased trial preparation time, decreased compensation expense for R&D employees, decreased manufacturing expenses, and decreased license expenses. In the latter months of the year ended December 31, 2020, patient enrollment costs increased as a result of the impact of the COVID-19 pandemic resulting in the opening of higher cost sites in Scandinavia as opposed lower cost sites in Central and Eastern Europe.
General and administrative expenses
General and administrative expenses increased by approximately $796,000 (19%) from in the year ended December 31, 2020 compared to the year ended December 31, 2019. This increase was due to increased expense in a several areas, including business development and financial consulting (approximately $141,000), public and shareholder relations (approximately $83,000), insurance expenses (approximately $46,000), and equity compensation expense (approximately $222,000) with the increased use of option and share grants.
Other income (expenses)
Other income increased by $382,000 (110%) in the year ended December 31, 2020 compared to the year ended December 31, 2019. This was mostly due to the presence in the first quarter of 2019 of a one-time warrant modification expense of $442,000, which was offset by a decrease in interest income of approximately $63,000, which was in turn due to the combination of substantially lower money market returns during the COVID-19 pandemic and somewhat reduced cash balances as we expend capital in the course of completing its trial.
Liquidity and capital resources
Overview
Our principal liquidity needs have historically been working capital, R&D, patent costs and personnel costs. We expect these needs to continue to increase in the near term as we develop and eventually commercialize our compound, if approved. Over the next several years, we expect to increase our R&D expenses as we undergo clinical trials to demonstrate the safety and efficacy of our lead product candidate and as we further develop product candidates acquired from Purnovate. To date, we have funded our operations primarily with the proceeds from our initial and secondary public offerings and our equity line, as well as other equity financings and the issuance of debt securities prior to that. On July 31, 2018, we closed our initial public offering.
On February 25, 2019, we closed a follow on a public offering pursuant to which we raised approximately $8.2 million net of underwriting discounts and commissions and estimated offering expenses.
On June 11, 2020, we concluded a registered direct offering of 2,820,000 shares of common stock and in a concurrent private placement the sale of warrants to purchase 2,115,000 shares of common stock at an exercise price of $2.00 per share. The shares of common stock and accompanying warrants were sold directly to the buyers at a combined at-the-market price of $1.85 for a share and three quarters warrant. Gross proceeds of the offering, totaled $5,217,000, which after offering expenses, resulted in net proceeds of $4,657,215.
On September 21, 2020, we concluded a private placement with a related party of 357,143 shares of common stock at an above-market price of $1.40 per share, for total proceeds of $500,000.
On September 30, 2020, warrants to purchase 225,000 shares of common stock an exercise price of $2.00 per share were exercised for a total cash payment of $450,000.
Our current cash and cash equivalents are not expected to be sufficient to fund operations for the twelve months from March 22, 2021, which is the date of the filing of our Annual Report on Form 10-K for the fiscal year ended December 31, 2020, based our current projections. We have recently executed an equity purchase agreement with Keystone Capital, LLC, which allows us to obtain depending on our stock’s market price and us meeting other conditions, up to $15 million in equity financing. We did not sell any shares of our common stock pursuant to the equity line with Keystone Capital during the year ended December 31, 2020; however, subsequent to the end of the year, we sold 1,007,296 shares of our common stock for gross proceeds of $2.35 million. We also received approximately $1.4 million in proceeds from exercise of warrants to purchase 712,500 shares of common stock for an exercise price of $2.00 per share.
We expect to use approximately $8.6 million in cash during the twelve months ended December 31, 2021 for both trial costs, other R&D project costs, and general corporate expenses. We expect to exhaust funds on hand in the fourth quarter of 2021, given our expected trial costs, other project costs, and costs of Company overhead, and will require approximately $3.3 million in additional funding to reach the current expected database lock in February of 2022. Our ability to access funds under the equity purchase agreement is dependent upon the market price of our common stock, so access to these funds is not guaranteed. If we are unable to access funds under the equity purchase agreement, we would need to raise funds through other sources. There is no assurance that such funds could be raised by that time on acceptable terms. Moreover, if our trial activities are significantly delayed due to the coronavirus pandemic, we would not be able to reach database lock with cash on hand even with receipt of the grants to which we have applied.
We will also require additional financing as we continue to execute our overall business strategy, including an estimated $20 million for a second phase three trial. Our liquidity may be negatively impacted as a result of research and development cost increases in addition to general economic and industry factors. We anticipate that, our future liquidity requirements will be funded through the incurrence of indebtedness, additional equity financings or a combination. In addition, we may raise additional funds through grants and/or corporate collaboration and licensing arrangements.
If we raise additional funds by issuing equity securities or convertible debt, our shareholders will experience dilution. Debt financing, if available, would result in increased fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our products, future revenue streams or product candidates or to grant licenses on terms that may not be favorable to us. We cannot be certain that additional funding will be available on acceptable terms, or at all. Any failure to raise capital in the future could have a negative impact on our financial condition and our ability to pursue our business strategies.
Cash flows
| | | For the Year Ended
December 31, | |
| (rounded to nearest thousand) | | 2020 | | | 2019 | |
| Provided by (used in) | | | | | | |
| Operating activities | | $ | (7,633,000 | ) | | $ | (6,339,000 | ) |
| Investing activities | | | (350,000 | ) | | | – | |
| Financing activities | | | 5,607,000 | | | | 9,247,000 | |
| Net increase (decrease) in cash and cash equivalents | | $ | (2,376,000 | ) | | $ | 2,908,000 | |
Net cash used in operating activities
Net cash used in operating activities for the year ended December 31, 2020 consists primarily of net loss adjusted for certain non-cash items (including amortization and share-based compensation), and the effect of changes in working capital and other activities. The increase in cash used in operating activities ($1,294,000) for the year ended December 31, 2020 as compared to the year ended December 31, 2019, despite the larger increase in net loss between the same periods (of approximately $2,301,000), is due to the larger proportion of costs being non-cash costs, such as equity compensation expense, and to a large increase in accrued expenses in the year ended December 31, 2020 compared to the year ended December 31, 2019. Non-cash assets, such as pre-paid research and development, also increased substantially (by about $162,000) in the year ended December 31, 2020, compared to the year ended December 31, 2019.
Net cash provided by investing activities
Net cash provided by investing activities during the year ended December 31, 2020 consists of the $350,000 loan that we made to Purnovate prior to the closing of the Acquisition to be used for further research and development efforts.
Net cash provided by financing activities
Net cash provided by financing activities during the years ended December 31, 2020 and 2019 primarily consist of capital raising activities through debt and equity financing. Net cash provided by financing activities decreased $3,640,000 during the year ended December 31, 2020, and was primarily attributable to the lesser net proceeds of the registered direct offering, private placement, and warrant exercises compared to the public offering completed in 2019.
Off-balance sheet arrangements
We do not have any off-balance sheet arrangements.
Recent Accounting Pronouncements
See Note 3 to the financial statements for a discussion of recent accounting pronouncements.
Critical accounting policies and estimates
The preparation of the financial statements requires us to make assumptions, estimates and judgments that affect the reported amounts of assets and liabilities, the disclosures of contingent assets and liabilities as of the date of the financial statements, our expected liquidity needs and expected future cash positions, and the reported amounts of sales and expenses during the reporting periods. Certain of our more critical accounting policies require the application of significant judgment by management in selecting the appropriate assumptions for calculating financial estimates. By their nature, these judgments are subject to an inherent degree of uncertainty. On an ongoing basis, we evaluate our judgments, including those related to prepaid research and development, accruals associated with third party providers supporting clinical trials, realization of income tax assets, as well as the, fair value of stock based compensation to employees and service providers. We use historical experience and other assumptions as the basis for our judgments and making these estimates. Because future events and their effects cannot be determined with precision, actual results could differ significantly from these estimates. Any changes in those estimates will be reflected in our financial statements as they occur.
While our significant accounting policies are more fully described in Note 3 to our financial statements included elsewhere in this prospectus, we believe that the following accounting policies and estimates are most critical to a full understanding and evaluation of our reported financial results.
R&D Expenses
Recognition and accrual of expenses associated with our clinical trial are dependent on the judgment of our contractors and subcontractors in their reporting and communication of information to us. Occurrence of certain fees to our CRO, clinical trial sites, and subcontractors are tied to events, for which the determination of likelihood requires judgment both on our part and on the part of our contractors.
Fair Value of Financial Instruments and Fair Value Measurements
Our financial instruments consist primarily of cash, accounts payable and accrued liabilities, and, prior to our initial public offering, debt instruments and derivative liabilities.
FASB Accounting Standards Codification (“ASC”) Topic 820, “Fair Value Measurements and Disclosures,” requires disclosure of the fair value of financial instruments held by us. ASC Topic 825, “Financial Instruments,” defines fair value, and establishes a three-level valuation hierarchy for disclosures of fair value measurement that enhances disclosure requirements for fair value measures. The carrying amounts reported in the balance sheets for receivables, current liabilities, convertible notes, payable senior notes, and bridge notes each qualify as financial instruments and are a reasonable estimate of their fair values because of the short period of time between the origination of such instruments and their expected realization and their current market rate of interest.
The three levels of valuation hierarchy are defined as follows:
| | ● | Level 1: Observable inputs such as quoted prices in active markets; |
| | ● | Level 2: Inputs, other than the quoted prices in active markets, that are observable either directly or indirectly; and |
| | ● | Level 3: Unobservable inputs in which there is little or no market data, which require the reporting entity to develop its own assumptions. As of December 31, 2020, the significant inputs to our derivative liabilities recorded at fair value were considered level 3 inputs. |
Stock Based Compensation
We estimate the fair value of options and stock warrants granted using the Black Scholes Merton model. We estimate when and if performance-based awards will be earned. If an award is not considered probable of being earned, no amount of equity-based compensation expense is recognized. If the award is deemed probable of being earned, related equity-based compensation expense is recorded. The fair value of an award ultimately expected to vest is recognized as an expense, net of forfeitures, over the requisite service periods in our statements of operations, which is generally the vesting period of the award.
The Black Scholes Merton model requires the input of certain subjective assumptions and the application of judgment in determining the fair value of the awards. The most significant assumptions and judgments include the expected volatility, risk-free interest rate, the expected dividend yield, and the expected term of the awards. In addition, the recognition of equity-based compensation expense is impacted by our forfeitures, which are accounted for as they occur.
The assumptions used in our option pricing model represent management’s best estimates. If factors change and different assumptions are used, our equity-based compensation expense could be materially different in the future. The key assumptions included in the model are as follows:
| | ● | Expected volatility — We determine the expected price volatility based on the historical volatilities of a peer group as we do not have a sufficient trading history for our units. Industry peers consist of several public companies in the bio-tech industry similar to us in size, stage of life cycle and financial leverage. We intend to continue to consistently apply this process using the same or similar public companies until a sufficient amount of historical information regarding the volatility of our own stock price becomes available, or unless circumstances change such that the identified companies are no longer similar to us, in which case, more suitable companies whose share prices are publicly available would be utilized in the calculation. Starting in 2020, we have begun blending data on the our historical volatility together with this peer group of companies, the proportion of our volatility used growing as the period of our historical volatility becomes longer. |
| | ● | Risk-free interest rate — The risk free rate was determined based on yields of U.S. Treasury Bonds of comparable terms. |
| | ● | Expected dividend yield — We have not previously issued dividends and do not anticipate paying dividends in the foreseeable future. Therefore, we used a dividend rate of zero based on our expectation of additional dividends. |
| | ● | Expected term —The expected term of the options was estimated using the simplified method. |
Commitments and Contingencies
We follow subtopic 450-20 of the FASB Accounting Standards Codification to report accounting for contingencies. Certain conditions may exist as of the date the financial statements are issued, which may result in a loss to us but which will only be resolved when one or more future events occur or fail to occur. We assess such contingent liabilities, and such assessment inherently involves an exercise of judgment.
If the assessment of a contingency indicates that it is probable that a material loss has been incurred and the amount of the liability can be estimated, then the estimated liability would be accrued in our financial statements. If the assessment indicates that a potentially material loss contingency is not probable but is reasonably possible, or is probable but cannot be estimated, then the nature of the contingent liability, and an estimate of the range of possible losses, if determinable and material, would be disclosed.
Loss contingencies considered remote are generally not disclosed unless they involve guarantees, in which case the guarantees would be disclosed. Our legal costs associated with contingent liabilities are recorded to expense as incurred.
Income taxes
We account for income taxes using the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax basis and tax carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Deferred tax assets are reduced by a valuation allowance, if, based on the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. We have no history of being able to generate a profit, and no certainty as to our ability to do so in the future.
Implications of Being an Emerging Growth Company
We are an “emerging growth company,” as defined in the JOBS Act, and therefore we intend to take advantage of certain exemptions from various public company reporting requirements, including not being required to have our internal controls over financial reporting audited by our independent registered public accounting firm pursuant to Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and any golden parachute payments. We may take advantage of these exemptions until we are no longer an “emerging growth company.” In addition, the JOBS Act provides that an “emerging growth company” can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have elected to use the extended transition period for complying with new or revised accounting standards under the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates. We will remain an “emerging growth company” until the earlier of (1) the last day of the fiscal year: (a) following the fifth anniversary of the completion of our initial public offering; (b) in which we have total annual gross revenue of at least $1.07 billion; or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeded $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period. References herein to “emerging growth company” have the meaning associated with that term in the JOBS Act.
BUSINESS
Overview
We are a clinical-stage biopharmaceutical company focused on the development of therapeutics for the treatment or prevention of addiction and related disorders. Our lead investigational new drug product, AD04, is being developed as a therapeutic agent for the treatment of alcohol use disorder (“AUD”). In January 2021, we expanded our portfolio in the field of addiction with the acquisition of Purnovate, LLC, and we continue to explore opportunities to expand our portfolio in the field of addiction and related disorders, both through internal development and through acquisitions. Our vision is to create the world’s leading addiction focused pharmaceutical company.
AUD is characterized by an urge to consume alcohol and an inability to control the levels of consumption. We have commenced the landmark ONWARD™ pivotal Phase 3 clinical trial using AD04 for the potential treatment of AUD in subjects with certain target genotypes. As of this filing, all 25 planned clinical sites were actively enrolling patients, and the ONWARD trial was more than 50% enrolled. The trial is expected to be completed by the first quarter of 2022. We believe our approach is unique in that it targets the serotonin system and individualizes the treatment of AUD, through the use of genetic screening (i.e., a companion diagnostic genetic biomarker). We have created an investigational companion diagnostic biomarker test for the genetic screening of patients with certain biomarkers that, as reported in the American Journal of Psychiatry (Johnson, et. al. 2011 & 2013), we believe will benefit from treatment with AD04. Our strategy is to integrate the pre-treatment genetic screening into AD04’s label to create a patient-specific treatment in one integrated therapeutic offering. Our goal is to develop a genetically targeted, effective and safe product candidate to treat AUD by reducing or eliminating the patients’ consumption of alcohol.
We have a worldwide, exclusive license from the University of Virginia Patent Foundation (d.b.a the Licensing & Venture Group) (“UVA LVG”), which is the licensing arm of the University of Virginia, to commercialize our investigational drug candidate, AD04, subject to Food and Drug Administration (“FDA”) approval of the product, based upon three separate patent application families, with patents issued in over 40 jurisdictions, including three issued patents in the U.S. Our investigational agent has been used in several investigator-sponsored trials and we possess or have rights to use toxicology, pharmacokinetic and other preclinical and clinical data that supports our landmark ONWARD pivotal Phase 3 clinical trial. Our therapeutic agent was the product candidate used in a University of Virginia investigator sponsored Phase 2b clinical trial of 283 patients. In this Phase 2b clinical trial, ultra-low dose ondansetron, the active pharmaceutical agent in AD04, showed a statistically significant difference between ondansetron and placebo for both the primary endpoint and secondary endpoint, which were reduction in severity of drinking measured in drinks per drinking day (1.71 drinks/drinking day; p=0.0042), and reduction in frequency of drinking measured in days of abstinence/no drinking (11.56%; p=0.0352), respectively. Additionally, and importantly, the Phase 2b results showed a significant decrease in the percentage of heavy drinking days (11.08%; p=0.0445) with a “heavy drinking day” defined as a day with four (4) or more alcoholic drinks for women or five (5) or more alcoholic drinks for men consumed in the same day.
The active pharmaceutical agent in AD04, our lead investigational new drug product, is ondansetron, which is also the active ingredient in Zofran®, which was granted FDA approval in 1991 for nausea and vomiting post-operatively and after chemotherapy or radiation treatment and is now commercially available in generic form. In studies of Zofran®, conducted as part of its FDA review process, ondansetron was given acutely at dosages up to almost 100 times the dosage expected to be formulated in AD04 with the highest doses of Zofran® given intravenously (“i.v.”), which results in approximately 160% of the exposure level as oral dosing. Even at high doses given i.v. the studies found that ondansetron is well-tolerated and results in few adverse side effects at the currently marketed doses, which reach more than 80 times the AD04 dose and are given i.v. The formulation dosage of ondansetron used in our drug candidate (and expected to be used by us in our Phase 3 clinical trials) has the potential advantage that it contains a much lower concentration of ondansetron than the generic formulation/dosage that has been used in prior clinical trials, is dosed orally, and is available with use of a companion diagnostic genetic biomarker. Our development plan for AD04 is designed to demonstrate both the efficacy of AD04 in the genetically targeted population and the safety of ondansetron when administered chronically at the AD04 dosage. However, to the best of our knowledge, no comprehensive clinical study has been performed to date that has evaluated the safety profile of ondansetron at any dosage for long-term use as anticipated in our ongoing and planned clinical trials.
According to the National Institute of Alcohol Abuse and Alcoholism (the “NIAAA”) and the Journal of the American Medical Association (“JAMA”), in the United States alone, approximately 35 million people each year have AUD (such number is based upon the 2012 data provided in Grant et. al. the JAMA 2015 publication and has been adjusted to reflect a compound annual growth rate of 1.13%, which is the growth rate reported by U.S. Census Bureau for the general adult population from 2012-2017), resulting in significant health, social and financial costs with excessive alcohol use being the third leading cause of preventable death and is responsible for 31% of driving fatalities in the United States (NIAAA Alcohol Facts & Statistics). AUD contributes to over 200 different diseases and 10% of children live with a person that has an alcohol problem. According to the American Society of Clinical Oncologists, 5-6% of new cancers and cancer deaths globally are directly attributable to alcohol. And, The Lancet published that alcohol is the leading cause of death in people ages 15-49 globally. The Centers for Disease Control (the “CDC”) has reported that AUD costs the U.S. economy about $250 billion annually, with heavy drinking accounting for greater than 75% of the social and health related costs. Despite this, according to the article in the JAMA 2015 publication, only 7.7% of patients (i.e., approximately 2.7 million people) with AUD are estimated to have been treated in any way and only 3.6% by a physician (i.e., approximately 1.3 million people). In addition, according to the JAMA 2017 publication, the problem in the United States appears to be growing with almost a 50% increase in AUD prevalence between 2002 and 2013.
AUD is characterized by an urge to consume alcohol and an inability to control the levels of consumption. Until the publication of the fifth revision of the Diagnostic and Statistical Manual of Mental Disorders in 2013 (the “DSM-5”), AUD was broken into “alcohol dependence” and “alcohol abuse”. More broadly, overdrinking due to the inability to moderate drinking is called alcohol addiction and is often called “alcoholism”, sometimes pejoratively.
Since ondansetron is already manufactured for generic sale, the active ingredient for AD04 is readily available from several manufacturers, and we have contracted with a U.S. manufacturer to acquire ondansetron at a cost expected to be under $0.01 per dose. Clinical trial material (“CTM”) has already been manufactured for the ONWARD Phase 3 trial. The CTM has demonstrated good stability after four years with the stability studies to date.
We have also developed the manufacturing process at a third-party vendor to produce tablets at what we expect will serve for commercial scale production (i.e., greater than 1 million tablets per batch), also at a cost expected to be less than $0.01 per dose. A proprietary packaging process has been developed, which appears to extend the stability of the drug product. Packaging costs are expected to be less than $0.05 per dose. We do not have a written commitment for supply of either the tablets or the packaging and believe that alternative suppliers are available to whom we can transfer the processes that have been developed.
Methods for the companion diagnostic genetic test have been developed as a blood test, and we established the test with a third-party vendor capable of supporting the ONWARD Phase 3 clinical trial. Additionally, we have built validation and possible approval of the companion diagnostic into the Phase 3 program, including that we plan to store blood samples for all patients in the event additional genetic testing is required by regulatory authorities.
Recent Developments
Clinical Developments
In January 2020, we announced that we had received favorable opinions from the Finnish Medicines Agency (FIMEA) and National Committee on Medical Research Ethics (TUKIJA) to commence our landmark ONWARD pivotal Phase 3 clinical trial to investigate AD04 as a genetically targeted therapeutic agent for the treatment of AUD.
On June 11, 2020, we announced that we had received all necessary approvals to commence our landmark ONWARD pivotal Phase 3 clinical trial to investigate AD04 as a genetically targeted therapeutic agent for the treatment of AUD.
On July 17, 2020, we received notice that the European Medicines Agency (EMA) had accepted the Pediatric Investigation Plan (PIP) submitted by us for development of our lead drug candidate, AD04, for the treatment of alcohol use disorder in the pediatric population, ages 12 to 17.
On July 30, 2020, we announced that we had received approval to commence our landmark ONWARD pivotal Phase 3 clinical trial of AD04 for the treatment of AUD in Croatia. As a result, we secured approvals to run the trial in all of the Scandinavian and Eastern European countries in which we intend to run the clinical trial.
On September 14, 2020, we filed with the FDA to take our Investigational New Drug (IND) Application for the use of our lead product candidate, AD04, as a treatment for Alcohol Use Disorder (AUD) off inactive status with the United States Food and Drug Administration (FDA) and on October 15, 2020 we announced that that the FDA had reactivated the IND.
On December 31, 2020, we provided the following highlighted updates on our landmark ONWARD pivotal Phase 3 clinical trial of AD04 for the treatment of AUD
| | ● | All 25 planned investigative sites are active. |
| | ● | 35% of the planned 290 subjects in the ONWARD trial were enrolled |
| | ● | The enrollment rate has increased for four consecutive months and the early termination rate has been lower than expected. |
| | ● | AD04 has been well tolerated with no serious adverse events having been reported |
Acquisition of Purnovate, LLC
On January 26, 2021, we closed the acquisition (the “Acquisition”) contemplated by that certain Equity Purchase Agreement, dated December 7, 2020, as amended (the “Purchase Agreement”), by and among Adial, Purnovate, LLC (“Purnovate”), each of the members of Purnovate (the “Members”) and Dr. Robert D. Thompson, as representative of the Members.
Prior to closing, we had advanced Purnovate $350,000 for use as working capital during the due diligence period. At closing, in exchange for Purnovate, Adial paid the members an additional $350,000 (the “Cash Consideration”) and issued to the members an aggregate of 700,000 shares of Adial restricted common stock (the “Stock Consideration”) with an approximate total market value of $1,638,000. In addition, members will receive (i) development milestone payments in an aggregate amount of up to $2,100,000 for each compound developed, (ii) development milestone payments in an aggregate amount of up to $20,000,000 for each compound commercialized, and (iii) royalties of 3.0% of Net Sales (as such term is defined in the Purchase Agreement). The Stock Consideration has been placed into escrow to secure certain indemnification and other obligations of Purnovate and the members in connection with the Acquisition.
The acquisition was effected by a merger (the “Merger”) of Purnovate into Purnovate, Inc., a Delaware corporation and wholly owned subsidiary of Adial, which survived the Merger. In connection with the Merger, on January 20, 2021, Purnovate converted from a limited liability company to a corporation and on January 25, 2021, the parties entered into an Amendment to the Purchase Agreement (the “Amendment”) to provide for the mechanism of closing the Acquisition through a Merger.
Purnovate is a drug development company with a platform focused on developing drug candidates for non-opioid pain reduction and other diseases and disorders potentially targeted with adenosine analogs that are selective, potent, stable, and soluble. Purines are a class of chemical structures that include adenosine, an important neurotransmitter. Purnovate uses innovative methods and technologies to enhance the drug properties of purines, which are a class of chemical structures that include adenosine, an important neurotransmitter. Adenosine receptors are hypothesized to be involved in the mediation of nociception (i.e., pain) and blocking the receptor is proposed as an anti-nociceptive. Additionally, selective adenosine analogs may be useful to treat pain while avoiding the tolerability problems (i.e., wakefulness, gastrointestinal) and safety issues (e.g., cardiac block and vasodilation) and therefore have the potential to provide a meaningful non-opioid treatment for pain without the side effects of earlier generations of adenosine compounds or certain other pain products. These adenosine analogs may also be useful in a number of other diseases and disorders such as cocaine addiction, infectious disease, inflammation, cancer, asthma, and diabetes. Purnovate’s portfolio of pre-clinical technologies are also believed to offer opportunities to improve the characteristics of other classes of molecules outside of the adenosine chemistry space and maybe even outside the purine chemistry space. All drug candidates developed using Purnovate’s platform technologies are expected to be patently distinct new chemical entities (i.e., patentable compositions of matter). Purnovate operates a chemistry and analytics laboratory in its 4,175 square feet leased laboratory and office space. Purnovate has been synthesizing new adenosine analog chemical entities with promising potency, selectivity, stability, and solubility characteristics.
Dr. Thompson, Purnovate’s Chief Executive Officer who continued his employment with Purnovate and joined Adial as its Vice President of Chemistry after the Acquisition, is a distinguished adenosine chemist that has been working in the field for over 35 years. He is an inventor on over 20 adenosine analog patents covering tens of thousands of novel molecules and has authored dozens of scientific publications.
Purnovate, was formed in 2019 by Dr. Thompson, William Stilley, Chief Executive Officer of Adial and a Member, Mikel Poulsen, a Member and consultant to Purnovate, and Cameron Black, a Member.
The Stock Consideration has been placed into escrow to secure certain indemnification and other obligations of Purnovate and the equity holders in connection with the Acquisition:
| | (i) | with respect to the equity holders other than William Stilley and Dr. Thompson, five (5) days after the effective date of a registration statement registering such shares with respect to thirty percent (30%) of such shares to be received by such equity holders and on the one (1) year anniversary of the closing with respect to seventy percent (70%) of such shares to be received by such equity holders; |
| | (ii) | with respect to Dr. Thompson, five (5) days after the effective date of a registration statement registering such shares with respect to thirty percent (30%) of such shares to be received by him; on the one (1) year anniversary of the closing with respect to twenty percent (20%) of such shares to be received by him; and with respect to remaining fifty percent (50%) of such shares to be received by him; on the earlier of the two (2) year anniversary of the closing or on the termination date of his employment if termination is by us without cause; and |
| | (iii) | with respect to William Stilley, on the earlier of the two (2) year anniversary of the closing with respect to all of such shares to be received by him or on the termination date of his employment if termination is by us without cause. |
The Stock Consideration, if not used to satisfy indemnification obligations, and the cash consideration will be distributed to the equity holders on a pro rata basis based on each such equity holders’ equity interest in Purnovate as compared to the aggregate Purnovate equity interests held by all equity holders.
In addition to the payments described above, under the terms of the Equity Purchase Agreement, we agreed to make cash payments to Dr. Thompson, as representative of the members for the benefit of such members in an amount equal to (i) 3.0% of Net Sales (as such term is defined in the Purchase Agreement) and (ii) upon the achievement of the following development and commercialization milestones:
| Milestone Event | | Milestone Payment | |
| First human dosing | | $ | 300,000 | |
| First dose in a Phase 2 Trial | | $ | 300,000 | |
| First dose in a Phase 3 Trial | | $ | 400,000 | |
| First acceptance of U.S. NDA submission | | $ | 500,000 | |
| First acceptance of NDA equivalent submission in Europe | | $ | 300,000 | |
| First acceptance of NDA equivalent submission in Asia | | $ | 300,000 | |
| First Commercial Sale in the U.S. | | $ | 10,000,000 | |
| First Commercial Sale in Europe | | $ | 5,000,000 | |
| First Commercial Sale in Asia | | $ | 5,000,000 | |
| Total potential | | $ | 22,100,000 | |
The Equity Purchase Agreement contains customary representations, warranties and covenants of us, Purnovate and the equity holders. Subject to certain customary limitations, the members have agreed to indemnify us and our officers and directors against certain losses related to, among other things, breaches of Purnovate’s and the Members’ representations and warranties, certain specified liabilities and the failure to perform covenants or obligations under the Purchase Agreement.
In connection with the Acquisition, Dr. Thompson entered into an employment agreement with us and a lock-up agreement with a term of two (2) years with respect to fifty percent (50%) of the Stock Consideration received by him, or his termination of employment by us without cause, if earlier. William Stilley entered into a lock-up agreement with a term of two (2) years with respect to one hundred percent (100%) of the Stock Consideration received by him, or his respective termination of employment by us without cause, if earlier.
In connection with entering into the Purchase Agreement, we loaned Purnovate $350,000 to continue its research and development efforts, which loan is evidenced by a 3.5% promissory note due December 7, 2021.
William B. Stilley, our President and Chief Executive Officer and a member of its board of directors, and James W. Newman, a member of our board of directors, were members of Purnovate. In connection with the Acquisition Mr. Stilley sold approximately a 28.7% interest in Purnovate for 201,109 shares of Adial common stock and Mr. Newman, through two entities he controls, together sold an aggregate 0.53% interest in Purnovate for 3,731 shares of Adial common stock, which shares have been placed in escrow. Messrs. Stilley and Newman, through two entities he controls, also received their respective pro rata share of the cash consideration paid by us to the Members.
COVID-19 Impact
As we advance our clinical programs, we are in close contact with our CROs and clinical sites and are assessing the impact of COVID-19 on our studies and current timelines and costs. In order to protect our patients in the ONWARD Phase 3 trial and potentially increase retention rates, we secured distribution rights to certain SARS-CoV-2 antibody tests to administer to patients in the trial and various time points. In an effort to make the antibody tests more widely available in the United States, we entered engagements with third parties for sell tests provided by Adial as their distributor. Significant resources have not been devoted to this effort and the impact on the Company is not expected to be material.
Disease Targets and Markets
Limitations of Current AUD Therapies
Today the most common treatments for AUD are directed at achieving abstinence and typical treatments include psychological and social interventions. Most therapies actually require abstinence prior to initiating therapy. Abstinence requires dramatic lifestyle changes often with serious work and social consequences. Frequently, patients cannot attend family and social events in order to ensure compliance with abstinence, and patients often must suffer from the stigma of having been labelled an alcoholic. Significant side effects of current pharmacologic therapies include mental side effects such as psychiatric disorders and depressive symptoms and physical side effects such as nausea, dizziness, vomiting, abdominal pain, arthritis and joint fitness. In fact, according to peer reviewed studies referenced in The Sober Truth: Debunking the Bad Science Behind 12-Step Programs and the Rehab Industry, L. Dodes and Z. Dodes, 2014 by Dr. Lance Dodes, the former Director of the substance abuse treatment unit of Harvard’s McLean Hospital, 90% or more of patients that use current therapy solutions, such as Alcoholics Anonymous, do not achieve long-term abstinence.
There are four drugs approved by the FDA and marketed in the United States for the treatment of alcohol addiction, Antabuse ® (disulfram) Vivitrol ® (naltrexone), Revia ® (naltrexone) and Campral ® (acomprosate) and one drug, Selincro ® (nalmefene) is marketed outside of the United States. All of the approved drugs, other than Selincro ®, require abstinence prior to commencing treatment with the drug, and all five drugs are known to have significant side effects.
Antabuse ® was approved for the treatment of alcohol dependence more than 50 years ago, making it the oldest such drug on the market. It works by interfering with the body’s ability to process alcohol. Its method of action and purpose is to cause patients that drink alcohol while taking Antabuse ® to experience numerous and extremely unpleasant adverse effects, including, among others, flushing, nausea, and palpitations, with the goal that patients will continue the medication but refrain from drinking in order to avoid these effects.
Naltrexone, which can be taken as a once-daily pill (Revia®) or in an approved once-monthly injectable form (Vivitrol ® ) that requires a doctor to administer is often associated with gastrointestinal complaints and has been reported to cause liver damage when given at certain high doses. As a result, it carries an FDA boxed warning, a special emphasized warning, for this side effect.
Campral®, taken by mouth three times daily, acts on chemical messenger systems in the brain.
Selincro® has not been approved for sale in the United States.
Our Proposed Solution
Our goal with AD04 is to develop an effective and safe product to treat AUD that does not require abstinence as part of the treatment and does not have the negative side effects of the current drugs on the market. Our product candidate, AD04, is designed for genotype positive patients who desire to control their drinking but cannot or do not want to completely abstain from drinking. By removing the difficulties associated with abstinence and the side effects associated with the other current products on the market, we believe that we may be able to remove barriers to patient adoption that inhibit adoption of current therapies and can attract a greater portion of the many millions of patients with AUD that remain untreated. Unlike other therapies, our investigational product, AD04, uses a novel mode of action for treating AUD that involves genetic screening with a companion diagnostic genetic test prior to treatment and is designed to reduce cravings for alcohol to effectively curb alcohol intake, without the requirement of abstinence prior to or during treatment. Our product candidate is intended to be easy to use since it is administered orally, currently on a twice daily basis and with a once-a-day tablet planned as part of the product’s life cycle management. To date, clinical testing of AD04 has shown it to have a positive safety and tolerability profile with side effects similar to placebo.
The companion diagnostic genetic test to be used to identify patients that are most likely to benefit from treatment with AD04 may potentially enhance the likelihood of a successful outcome for those undergoing treatment. Additionally, it may provide doctors with the opportunity to have a non-threatening conversation about alcohol with their patients and may provide the patient an acceptable path to help them determine if they might be a candidate for help with their alcohol use. If the test results are positive, they would have a science-based rationale for their treatment, which reduces some of the stigma patients might otherwise endure, and allows them to be treated in the confidence of their doctor, potentially with a simple, oral tablet.
Strengths and Competitive Advantages
Large Market Opportunity for an Effective Solution
In the United States alone, approximately 35 million people each year have AUD. Based on data from the Phase 2b trial of AD04 and our analysis of publicly available genetic databases, we preliminarily estimate that about one in three patients with AUD in the U.S. will have the genetic markers to indicate possible treatment with AD04. At this time, we are not aware of any oral pharmaceutical treatment approved in the U.S. that addresses the needs of patients who desire to control their drinking but cannot or do not want to abstain from drinking. The current abstinence-based treatments have limitations. The limited side effects expected for our investigational new drug, based on clinical data so far, are also believed to be an important factor in the expected rapid uptake of AD04 in the market. Our approach, if approved by FDA, may allow for social drinking to continue and is aimed at reducing the dangerous, heavy drinking. This would allow patients to live the life they want without the stigma associated with complete abstention and currently endured by those seeking help for their excessive drinking. Assuming that one-third of AUD patients are genotype positive for treatment with AD04 and a $255 price for a one month supply of the drug (assumed pricing based on an average of prices published by Blue Cross Blue Shield in June 2017 for tier-3 oral, on-patent, chronic maintenance drugs, discounted by 16.6%, to reflect the average difference between retail and wholesale pricing for branded drugs as reported by drugs.com), the total potential market for AD04 would be approximately $36 billion in the United States alone.
Beyond the United States, alcohol consumption worldwide is a serious health issue. The 2014 Global Status Report on Alcohol and Health published by the World Health Organization (the “WHO”) states that 5.9% of all deaths (about 3.3 million per year) and 5.1% of disease worldwide are attributable to alcohol consumption. Europe consumes over 25% of the total alcohol consumed worldwide despite only having 14.7% of the world’s population. The WHO estimates that about 55 million people in Europe have AUD and, within Europe, Eastern Europe has a particularly acute problem with Russia estimated to have about 21 million people with AUD. The WHO further estimates that 17.4% of adult Russians and 31% of adult Russian males have AUD, and the Organization for Economic Cooperation and Development data indicates that 30% of all deaths in Russia are alcohol related as reported by Quartz Media.
Companion Genetic Bio-Marker Aimed at Identifying Patients Most Likely to Respond To Treatment, Potentially Results in Increased Use of AD04
We believe our drug is unique in that it is designed to reduce heavy drinking in individuals with certain genotypes. We are pursuing a strategy that aims to integrate pre-treatment screening with the companion diagnostic genetic test into the drug label, essentially combining the test and treatment into one integrated therapeutic offering that has combined intellectual property protections. This companion diagnostic testing approach may be a useful genetic screening tool to predict those most likely to respond to the drug and to have minimal side effects. Based on the clinical experience to date and publicly available databases, we believe the genetic prevalence of genotype positive people is about 33% of the population in the United States and that the prevalence in Scandinavia and in certain areas of Central and Eastern Europe may be greater than 50%. The FDA has agreed that the Phase 3 trials of AD04 can proceed only enrolling patients that are genotype positive, which greatly reduces the cost, time and risk relative to a trial that also enrolled patients that are genotype negative for treatment with AD04. We are conducting our current landmark ONWARD pivotal Phase 3 clinical trial in counties in Scandinavia and Central and Eastern Europe, including Finland, Sweden, Estonia, Latvia, Poland, Bulgaria, and Croatia. We expect to use the ONWARD trial as a pivotal Phase 3 trial to serve as a basis for approval in both the United States and Europe.
We believe that the companion diagnostic genetic test enables physicians to more easily have an initial conversation with their patients about alcohol use and, for the patient, provides a less threatening and obtrusive first step toward treatment because the conversation will include the topic of genetic testing and not be solely about behavior. Patients that then test positive against the AD04 genetic panel would be expected to be more likely to then receive a prescription for AD04 (based on an external quantitative market study of 156 primary care physicians and psychiatrists that was conducted by Ipsos-Insight LLC, who we commissioned, and that concluded a majority of genetically targeted patients currently receiving pharmacologic treatment would be switched to a drug with the characteristics expected for AD04).
Prior Work of Universities and our Ability to Leverage Relationships Creates Cost Efficiencies
We have a worldwide, exclusive license to intellectual property developed at the University of Virginia by our Chief Medical Officer and largest stockholder, Dr. Bankole A. Johnson, who was Chairman of the Department of Psychiatry & Neurobehavioral Sciences at the University of Virginia (and prior to that the Chief of the Division of Alcohol and Drug Addiction at the University of Texas) and was Chair, Department of Psychiatry and Director of the Brain Science Research Consortium Unit at the University of Maryland. Dr. Johnson has spent almost three decades researching the underlying subject matter. Significant portions of the supporting research were also funded under grants from the National Institute of Health to the University of Virginia and the University of Texas. On July 5, 2019, we entered into a Master Services Agreement and statement of work with Psychological Education Publishing Company (“PEPCO”), a company owned by Dr. Johnson, that is engaged in the business of administering a behavioral therapy program, Brief Behavioral Compliance Enhancement Treatment, for our upcoming Phase 3 clinical trial using AD04, for the treatment of AUD.
By leveraging the prior work of universities and their researchers, including their pre-clinical studies and accumulated data, we believe we have developed a significant drug development opportunity. Because of the licensing approach taken to secure intellectual property, including, without limitation, patents and rights to clinical trial data, and our collaborations with the University of Virginia, we, historically have not had to incur the significant costs that would normally be required to develop therapeutic treatments to the point of being ready to commence a Phase 3 clinical trial, which often amount to tens of millions of dollars or more. In fact, based upon current information, and depending on what the regulatory authorities may require to secure marketing authorization, we estimate that we will require approximately $10.7 million for the current Phase 3 clinical trial (not including company overhead) and an additional $30 million of additional capital to complete our second Phase 3 program (which includes $20 million for a confirmatory Phase 3 trial and any necessary Phase 1 clinical trials and other development expenses and does not include the additional cost of a possible third Phase 3 clinical trial) as currently contemplated in order to achieve regulatory approval for the use of AD04 to treat AUD in the United States and Europe. We have already used approximately $5.3 million in funds derived from our initial public offering and subsequent financings and warrant exercises to fund trial activities. We anticipate that the approximate $5.4 million needed to complete the initial Phase 3 clinical trial to the point of achieving database lock will be funded from our cash on hand, funds from additional financings, grant awards, and/or funds received from the exercise of warrants. We anticipate, with our expected rate of expenditure, to have exhausted our funds on hand by the end of November of 2021. We have entered into an equity purchase agreement with Keystone Capital, LLC, which provides for the sale of up to $15 million in common stock, of which $13 million remains available at the time of filing. These funds would be more than sufficient to fund our operations over the coming year and to completion of the initial Phase 3 trial. However, as our ability to access these funds is dependent upon the market price of our common stock, access to these funds is not guaranteed. If we are unable to access these funds, we may have to obtain additional funds through other means. There is no assurance that such funds could be raised in time to complete the trial on acceptable terms.
The NIAAA has provided and continues to provide technical assistance and advice to us, and we have applied for an NIAAA Research Resource Award, which if granted would provide financial support for our Phase 3 clinical trial. Although there can be no assurance that we will be selected by the NIAAA to receive funding, since we are not aware of any pharmaceutical company planning Phase 3 pivotal trials to serve as a basis for marketing approval for products for the treatment of AUD, we believe AD04 would be a competitive candidate. Currently, much of the funding expected for grants such as those for which we have applied has been diverted to COVID-19-related grants, and we are not certain if and when funding for grants such as ours will be available.
Known, Well-Tested Agent Has Shown Favorable Results in Non-AUD Uses
Ondansetron, the principal active pharmaceutical agent in AD04 has been approved by the FDA to treat nausea and vomiting but is administered at much higher doses than we intend to use and has shown limited side effects even at the higher dosages currently on the market. However, it has not been approved in our anticipated dosage or for our anticipated uses and treatment period. Consequently, we expect to submit a new drug application, pursuant to section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act, for U.S. marketing authorization. Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act allows the FDA to rely, for approval of an NDA, on data not developed by the applicant. Such an NDA contains full reports of investigations of safety and effectiveness, but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Such applications permit approval of applications other than those for duplicate products and permits reliance for such approvals on literature or an FDA finding of safety and/or effectiveness for an approved drug product. A Phase 2b University of Virginia investigator sponsored clinical trial of AD04 for the treatment of AUD showed promising results and no overt safety concerns (there were no statistically significant serious adverse events reported). Not only did the trial show no statistically significant, serious adverse side effects, but both of the pre-specified endpoints, reduction in severity of drinking measured in drinks per day of drinking day and reduction in frequency of drinking measured in days of abstinence, were met with statistical significance as shown in the graph below:
Phase 2b Clinical Trial Results– Analysis of Primary and Secondary Efficacy Endpoints for Target Genotypes
A 12-week, randomized, two-center, parallel-group, double-blind, placebo-controlled, two-arm (four cell) clinical trial of oral ondansetron (n=283) conducted by University of Virginia
Our Substantial Proprietary Estate and Protection from Competition
We currently hold a worldwide, exclusive license to three (3) patent families that provide us with the ability to exclude potential competitors from practicing the claimed inventions, such as the use of ondansetron to treat any of the four (4) specified genotypes for AUD. Our licensed patent estate is expected to provide us patent protection through 2032 plus possible extensions. Ondansetron, the active ingredient in AD04, has never been approved in a low dosage near the AD04 dose of 0.33mg per tablet, and we believe our licensed patents will protect AD04 from any competitor that attempts to bring to market an ondansetron dose at or near the AD04 dose for treatment of patients having one or more of the four target genotypes.
We believe use of the currently marketed doses “off-label” will not be significant due to (i) the lack of demonstrated efficacy at currently marketed doses, (ii) potential safety concerns if the currently marketed doses are used chronically as is expected to be necessary for treating AUD, and (iii) cutting the smallest currently marketed dose into the 12 pieces that would be necessary to achieve the AD04 dose is deemed by us to be impractical and likely to result in inaccurate dosing.
Companion Genetic Bio-Marker Aimed at Identifying Patients Most Likely to Respond To Treatment, Potentially Results in Increased Use of AD04
We believe our drug is unique in that it is designed to treat individuals with certain genotypes. We are pursuing a strategy that aims to integrate pre-treatment screening with the companion diagnostic genetic test into the drug label, essentially combining the test and treatment into one integrated therapeutic offering that has combined intellectual property protections. This companion diagnostic testing approach may be a useful genetic screening tool to predict those most likely to respond to the drug and to have minimal side effects. Based on the clinical experience to date and publicly available databases, we believe the genetic prevalence of genotype positive people is about 33% of the population in the United States and that the prevalence in Scandinavia and in certain areas of Central and Eastern Europe may be greater than 50%. The FDA has agreed that the Phase 3 trials of AD04 can proceed only enrolling patients that are genotype positive, which greatly reduces, the cost, time and risk relative to a trial that also enrolled patients that are genotype negative for treatment with AD04. The FDA has indicated that any approval based on a trial only in genotype positive patients would result in labeling restricted to treating genotype positive patients.
We believe that the companion diagnostic genetic test enables physicians to more easily have an initial conversation with their patients about alcohol use and, for the patient, provides a less threatening and obtrusive first step toward treatment because the conversation will include the topic of genetic testing and not be solely about behavior. Patients that then test positive against the AD04 genetic panel would be expected to be more likely to then receive a prescription for AD04.
Experienced Leadership
Our management, advisors and board of directors have extensive experience in pharmaceutical development, the clinical trial and regulatory approval processes, drug commercialization, financing capital-intensive projects, and developing new markets for pharmaceutical agents. Members of our team have previously worked in senior management and senior officer positions, or led significant research initiatives at Gensia, Clinical Data, Shire, Viagene, New River Pharmaceuticals, Collateral Therapeutics, Indivior, Krystal Biotech, Adenosine Therapeutics, and the University of Virginia and University of Maryland in a broad range of therapeutic areas. Our management and board members have particular expertise in the science and development of addiction related drugs and bringing new drugs to the market.
Our Strategy
We develop pharmaceutical treatments for addictions and addictive disorders and related diseases and disorders. Our business strategy is to advance AD04, our lead investigational drug candidate, toward regulatory approval for alcohol addiction in the United States, the European Union, and then eventually other territories. We subsequently plan to develop label expansions into other indications (e.g., opioid use disorder, other drug addictions, obesity, smoking cessation, eating disorders and anxiety). Additionally, we are inventing and developing novel therapeutic agents at our chemistry facilities and seeking to acquire addiction related assets, particularly those expected to be synergistic with AD04 once it is marketed, if it is approved.
Our goals in executing this strategy are to keep capital requirements to a minimum, expedite product development, gain access to clinical research and manufacturing expertise that will advance product development, approval and eventual market uptake of our product, and rely on a well-defined and carefully executed intellectual property strategy in order to position our products with long-term, defensible, competitive advantages. Execution of this strategy may include seeking grant funding and funding from partners and collaborators when available on terms we believe to be favorable to us, and on which there is no guarantee will be available. In collaboration with our CRO, we have been and are working to adapt the implementation of our strategy in response to the ongoing coronavirus pandemic.
Our near-term strategy includes:
| | ● | Obtaining regulatory approval for our lead product in the United States and Europe. We have commenced our initial Phase 3 clinical trial for the treatment of AUD in Scandinavia and Central and Eastern Europe. If our initial Phase 3 clinical trial is successful, we expect to conduct a second, and possibly a third, Phase 3 clinical trial in the same areas but with additional clinical sites in the United States and Western Europe. |
| | ● | Prosecuting and expanding our intellectual property and product portfolio. We have acquired rights to a promising drug candidate and made a significant investment in the development of our licensed patent portfolio to protect our technologies and programs, and we intend to continue to do so. We have obtained exclusive rights to three different patent families directed to therapeutic methods related to our AD04 platform. These families include 3 issued U.S. patents, and at least one foreign equivalent patent covering AD04 issued in over 40 national jurisdictions, including most of Europe and Eurasia. Divisional and continuation applications to expand the coverage have also been filed in certain jurisdictions. Additionally, commencing in early 2021, we have an adenosine platform that has and is expected to continue to generate what we believe are patentable new chemical entities. We intend that further product portfolio expansions will be focused on promising addiction therapies and/or late-stage clinical assets. |
| | | |
| | ● | Evaluating the additional use of our product candidate in other indications. In addition to alcohol addiction, we plan to conduct exploratory work to investigate using AD04 as a potential treatment for opioid use disorder, gambling addiction, smoking cessation, obesity, and other addiction related disorders in which 5-HT3 antagonism may have a treatment effect. We believe we will be able to undertake this initial exploratory effort with minimal additional cash cost to our company through the use of academic partnerships, grants, human laboratory studies and/or non-clinical studies. We believe that, due to its hypothesized mechanism of action (i.e., the modulation of the serotonin system in patients that are genetically targeted based on the apparent sensitivity to such modulation, where the modulation appears to reduce cravings), AD04 has the potential to be used for the treatment of such other addictive disorders. To date, we have not discussed these potential uses with the FDA or any other regulatory bodies. |
| | ● | Maximizing commercial opportunity for our technology. AD04 targets large markets with significant unmet medical need. We intend to develop an extended release, once-a-day formulation of AD04 to enhance compliance and market appeal. |
| | | |
| | ● | Managing our business with efficiency and discipline. We believe we have efficiently utilized our capital and human resources to develop and acquire our product candidate and programs and create a broad intellectual property portfolio. We operate cross-functionally and are led by an experienced management team with backgrounds in developing product candidates. We use project management techniques to assist us in making disciplined strategic program decisions and to attempt to limit the risk profile of our product pipeline. |
The clinical development plan for AD04 can be described as a two-stage development strategy in which we expend limited resources to achieve the significant value inflection point of Phase 3 data in our primary indication of AUD. With a successful trial and the risk reduction associated with that success, we would then be ready to conduct the final trials to seek approval in the U.S. and Europe as shown below:
AD04 — Two-Stage Clinical Development Strategy — Conduct the Phase 3 clinical trials sequentially
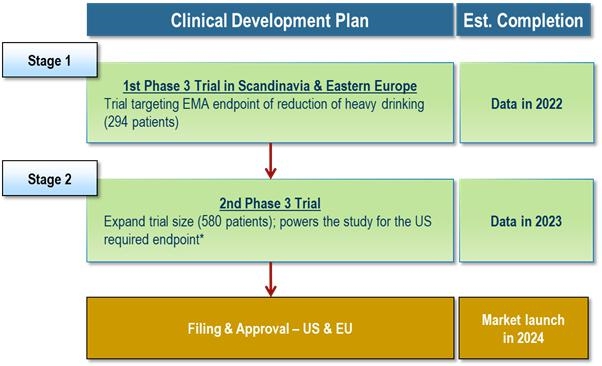
| * | Even if the 1st Phase 3 trial is not accepted by the FDA due to the study not being well-powered for the FDA’s currently stated end point, we still expect that the EMA will require only one additional trial. In this case, however, a 3rd trial might be required by the FDA (i.e., three Phase 3 trials in total). If two additional trials are required for FDA approval after an initial Phase 3 trial conducted in the EMA, we would expect to run the 2nd and 3rd trials in parallel (i.e., at the same time) so as not to increase the expected time to approval. The 2nd Phase 3 trial is expected to require $20 million in direct expenses, and up to $10 million in additional other development expenses is expected to be required. A possible 3rd Phase 3 trial would be expected to require an additional $20 million in clinical trial related expenditures. |
After approval, we plan to execute a two-stage commercialization plan for AD04. With psychiatrists and addiction specialists treating a majority of the current AUD patients today and with psychiatrists most likely to be familiar with the mechanism of action of AD04, we believe that a relatively small psychiatry-targeted, specialty sales force could successfully sell AD04 into the market. This plan creates the opportunity for us to develop into a commercial enterprise with an initial niche-market sales force at a relatively low cost for market entry. It also expands the universe of potential acquirers of our company or AD04 to smaller and mid-size pharmaceutical companies. Once success is shown in the niche market and the thought leaders and early adopters are prescribing AD04, market adoption risk will have been greatly reduced and we would expect to be able to sell or partner with a large pharmaceutical partner to develop AD04 as a blockbuster product. This commercialization plan is shown below:
AD04 — Two-Stage Commercialization Strategy — Initial launch with a specialty sales force to build the market, then partner or sell to a large pharmaceutical partner to capture market share and optimize the market
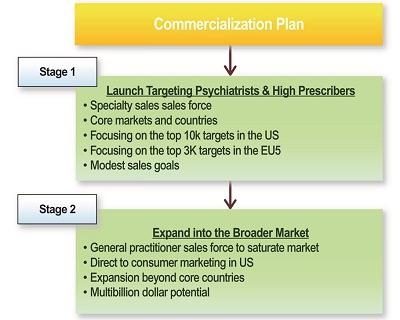
Ondansetron History and Foundation for Treating AUD
Ondansetron is a 5-HT3 receptor antagonist. Preclinical and pharmacobehavioral studies suggest that blockade of serotonin-3 receptors will influence the dopamine reward system activated by alcohol, decreasing dopamine release and attenuating craving for alcohol (Dawes, MA et al., 2005b; Johnson, BA et al., 2002; Lovinger, DM, 1999a). Early clinical studies found that the efficacy of ondansetron is limited to certain subgroups of the alcohol-dependent population and suggested the differential effect could be predicted based on age of onset of alcoholism, an indistinct concept likely confounded by genetic, regional and ethnic differences (Johnson, BA et al., 2000; Kranzler, HR et al., 2003). Recent research suggests the variable effect may be predictable based on molecular mechanism of ondansetron action and individual subject genotype of key genes in the serotonin system (Enoch, MA et al., 2010; Johnson, BA et al., 2011; Kenna, GA et al., 2009).
We are pursuing development of ondansetron in the alcohol-dependent population. Clinical studies will initially focus on the use of a low dose, oral tablet (0.33 mg administered twice daily) to reduce alcohol consumption in subjects with genotypes that have been correlated with a responsive to treatment with ondansetron.
Ondansetron was first approved by the FDA in 1991 as a solution for injection. Subsequent approvals were obtained for oral tablets in dosage forms and an oral solution. It is marketed as Zofran ® and is also available in generic formulations, and it has been used widely for the approved indications – prevention of nausea and vomiting associated with certain cancer chemotherapies and radiotherapies and for the prevention of postoperative nausea or vomiting — at adult doses of 8–24 mg/day with manageable side effects.
Ondansetron has been administered to dogs‚ rats‚ and mice as part of a preclinical toxicology program which included single-dose acute‚ repeated-dose studies. Ondansetron was not mutagenic in the standard battery of microbial tests for mutagenicity and no carcinogenic effects were seen in 2-year studies in rats and mice with oral ondansetron doses up to 10 and 30 mg/kg/day, respectively. In studies of rats and rabbits there was no evidence of reproductive toxicity seen on fertility, early embryonic development, perinatal/postnatal development or fetal development of the F2 generation. Based on these studies, as well as over 20 years of human use in clinical trials and the post-marketing environment, ondansetron is considered to be a well-tolerated drug with a generally mild safety profile.
Ondansetron, by blocking the 5-HT3 receptor, is known to affect dopaminergic signaling in the brain; and the scientific rational for use of a 5-HT3 antagonist in the treatment of alcohol dependence is well established (Johnson, BA, 2004). Briefly, studies suggest that: the rewarding effects of alcohol involve activation of the 5-HT3 receptors leading to release of dopamine within the mesolimbic system of the brain (McBride, WJ et al., 2004). Thus, by blocking activation of the 5-HT3 receptor, ondansetron may reduce the ethanol-stimulated release of dopamine leading to reduced feelings of pleasure or reward and consequently, reduced consumption (Carboni, E et al., 1989; Costall, B et al., 1987; Hagan, RM et al., 1990; Imperato, A and Angelucci, L, 1989; Lovinger, DM, 1999b; McBride, WJ et al., 2004; Minabe, Y et al., 1991; Rasmussen, K et al., 1991; Wozniak, KM et al., 1990; Yoshimoto, K et al., 1996).
Preclinical studies have demonstrated that alcohol stimulates the release of both serotonin (5-hydroxytryptamine or 5-HT) and dopamine within the cortio-mesolimbic system (Campbell, AD et al., 1996; Campbell, AD and McBride, WJ, 1995; Di Chiara, G and Imperato, A, 1988; Imperato, A and Angelucci, L, 1989; Yoshimoto, K et al., 1992; Yoshimoto, K et al., 1996; Zazpe, A et al., 1994). Other studies have shown that alcohol potentiates the effects of 5-HT at the 5-HT3 receptor, leading to augmented release of dopamine, and that ondansetron and the selective antagonists of the 5-HT3 receptor inhibit dopaminergic firing and release of dopamine in response to alcohol and serotonin (Costall, B et al., 1987; Lovinger, DM, 1991; Minabe, Y et al., 1991; Rasmussen, K et al., 1991; Yoshimoto, K et al., 1996; Zazpe, A et al., 1994; Zhou, Q et al., 1998). Finally, numerous in vivo studies in rats and mice have shown that ondansetron and other selective antagonist of the 5-HT3 receptor reduce volitional intake of alcohol in models selectively bred for alcohol preference (Fadda, F et al., 1991; Hodge, CW et al., 1993; McBride, WJ and Li, TK, 1998; Meert, TF, 1993; Tomkins, DM et al., 1995).
The aforementioned nonclinical studies have shown that 5-HT3 and dopamine interactions in the cortico-mesolimbic system appear to mediate many of the reinforcing effects of alcohol. Collectively the available nonclinical studies suggest that, by inhibiting the 5-HT3 receptor and reducing the release of dopamine in the cortico-mesolimbic area, ondansetron can interfere with the dopamine reward system activated by alcohol and lead to reduced alcohol intake (Barnes, NM and Sharp, T, 1999; Dawes, MA et al., 2005b; Johnson, BA et al., 1993; Johnson, BA and Cowen, PJ, 1993; Lovinger, DM, 1991, 1999a; Swift, RM et al., 1996; Tomkins, DM et al., 1995).
Five clinical studies have been conducted that demonstrate ondansetron is a promising treatment for alcohol-dependent individuals (Johnson, BA et al., 2011; Johnson, BA et al., 2000; Kenna, GA et al., 2009; Kranzler, HR et al., 2003; Sellers, EM et al., 1994). Several important findings in these studies guide the design of future clinical studies, including:
| | (1) | Ondansetron’s efficacy in alcohol-dependent individuals is associated optimally with a small dose of the compound (0.25-0.33 mg twice daily), a dose that is <1/10 of the dose used for adults for the currently approved indications. |
| | (2) | In clinical studies in over 600 subjects, ondansetron was well-tolerated and safe, with a mild side-effect profile when administered to currently drinking alcohol-dependent individuals. Overall, the types of adverse events reported during multi-week clinical studies in alcohol dependence appear similar to those outlined in the package insert for the approved indications and to those reported in the literature for treatment in chronic liver disease, chronic fatigue syndrome and schizophrenia. |
| | (3) | The extent of benefit with ondansetron treatment varies among different subtypes of alcohol-dependent subjects. Prior studies found that ondansetron benefited subjects with early-onset alcoholism (EOA) but not late-onset alcoholism (LOA). The pharmacological reason for this was not known, but it was presumed that the differential effect was due to a higher degree of serotonergic dysfunction in EOA (Johnson, BA et al., 2000; Kranzler, HR et al., 2003). |
The below table summarizes the five clinical studies demonstrating ondansetron is a promising treatment for alcohol-dependent individuals
| Study type (Reference) | | Number of
Subjects | | Dosing
(Duration) | | Summary Results |
Phase 2 (Sellers, EM et al., Clinical Efficacy of the 5-HT3 Antagonist Ondansetron in Alcohol Abuse and Dependence, Alcohol Clin Exp Res, 18 (1994) 879-885.) | | 71 | | 0.25 mg, 2 mg, and placebo b.i.d.
(6 weeks) | | The 0.25 mg dose showed a near significant effect in reducing severity of drinking measured in DDD (p=0.06) while the 2 mg dose was similar to placebo. |
| | | | | | | |
Phase 2 (Johnson, BA et al., Ondansetron for Reduction of Drinking among Biologically Predisposed Alcoholic Patients: A Randomized Controlled Trial, JAMA, 284 (2000) 963-971) | | 321 | | 1, 4, and 16 ug/kg b.i.d.
(11 weeks) | | Ondansetron treatment at doses of 1, 4, and 16 µg/kg bid resulted in significant reductions in DDD in EOA subjects, but only the 4 µg/kg dose showed such a reduction in frequency of drinking measured in PDA and the maximal effect was shown at the µg/kg does. Only the 4 µg/kg bid showed significant improvements in PDA in the LOA group. |
| | | | | | | |
Phase 2 (Kranzler, HR et al., A within-Group Design of Nontreatment Seeking 5-HTTLPR Genotyped Alcohol-Dependent Subjects Receiving Ondansetron and Sertraline, Alcohol Clin Exp Res, 33 (2009) 315-323) | | 40 | | 4 ug/kg bid for 8 weeks | | EOA subjects showed significant improvement over LOA subjects in DDD. |
| | | | | | | |
Phase 2 (Kenna, GA et al., Pharmacogenetic Approach at the Serotonin Transporter Gene as a Method of Reducing the Severity of Alcohol Drinking, Am J Psychiatry, 168 (2011) 265-275) | | 21 | | .5 mg/day for 3 weeks | | LL genotype subject showed significant improvement in DDD. |
| | | | | | | |
Phase 2b (Johnson, BA et al., Determination of Genotype Combinations That Can Predict the Outcome of the Treatment of Alcohol Dependence Using the 5-HT3 Antagonist Ondansetron, Am J Psychiatry (2013) | | 283 | | 4 ug/kg bid
(12 weeks, including 1 week placebo run-in) | | The target genotype group showed significant improvement in DDD and PDA against both the placebo groups and other genotypes on drug. |
Additional detail with respect to four of the clinical studies referenced in the chart above is provided below with the fifth being the Phase 2b clinical trial upon which we are basing the development of AD04 and which is described more fully in the following section titled “Phase 2b Investigator Initiated Clinical Trial of AD04 for Alcohol Use Disorder Conducted by the University of Virginia.”
A Dose-Ranging, Placebo-Controlled, 6-Week Study of Ondansetron in Alcoholic-Dependent Subjects
In 1994, Sellers et al. reported on the effects of administration of 0.25 mg bid ondansetron (N=23), 2 mg bid ondansetron (N=25), or placebo (N=23) for 6 weeks in alcohol-dependent males (Sellers, EM et al., 1994). Endpoints included change in drinks per drinking day (“DDD”) and proportion of responders, where a responder was defined as a subject with a Reliable Change score > 1.96, representing an improvement of at least 2 standard deviations. The Reliable Change score was calculated as the difference between pre- and post-test DDD divided by the standard error. Analyses were conducted comparing pre-treatment with the Week 6 visit, representing the end-of-study medication administration, and pre-treatment with the Week 7 visit, after completion of a 1-week follow-up period.
In the 71 subjects who completed the study, the on-treatment changes in DDD were approximately -1.9 (0.25 mg bid), -1.2 (2 mg bid), and -1.3 (placebo), with neither ondansetron effect being statistically different from the placebo effect. The corresponding changes from pre-treatment to Week 7 (after 6 weeks of treatment and a 1-week follow-up) were approximately -2.7 (0.25 mg bid), -1.1 (2 mg bid), and -1.6 (placebo), with the difference between low-dose ondansetron and placebo approaching statistical significance (p=0.06). By Week 6, nearly twice as many subjects on low-dose ondansetron compared with those on either high-dose ondansetron or placebo showed significant improvement according to the Reliable Change score. Lower baseline drinking and higher level of education were significant predictors of reduction in drinking while on treatment.
A Dose-Ranging, Placebo-Controlled, 11-Week Study of Ondansetron in Alcoholic-Dependent Subjects
In 2000, Johnson et al. reported on the co-administration of weekly cognitive behavioral therapy and either placebo or ondansetron at doses of 1, 4, and 16 µg/kg bid for 11 weeks (after a 1-week, single-blind, placebo lead-in) in 321 alcohol-dependent subjects (Johnson, BA et al., 2000). Endpoints included drinks per day, DDD, percentage of days abstinent (“PDA”), total days abstinent, and plasma carbohydrate deficient transferrin (CDT) level, an objective measure of drinking. Analyses were conducted comparing each dose group with placebo, with drinking response variables analyzed as means of data collected from Weeks 3 through 12.
The table below sets forth treatment results. Ondansetron treatment at doses of 1, 4, and 16 µg/kg bid resulted in statistically significant reductions in DDD and drinks per day compared with placebo for EOA (age of onset ≤25 years). The maximum clinical effect was observed at the middle dose (4 µg/kg bid), though the differences between doses were not statistically significant. At 4 µg/kg bid (but not at 1 or 16 µg/kg bid), significant improvements in days and PDA were also achieved. LOA (age of onset ≥26 years) did not benefit from ondansetron treatment at any dose studied.
Treatment Effect Size in EOA Subjects and Statistical Comparison to Placebo Effect
| Variable | | 1 µg/kg bid | | 4 µg/kg bid | | 16 µg/kg bid | |
| Drinks/drinking day | | 0.25 (p≤0.05) | | 0.41 (p≤0.01) | | 0.23 (p≤0.05) | |
| Drinks/day | | 0.26 (p≤0.05) | | 0.37 (p≤0.01) | | 0.22 (p≤0.05) | |
| Days abstinent (%) | | 0.13 (ns) | | 0.26 (p≤0.01) | | 0.17(ns) | |
| Days abstinent | | 0.06 (ns) | | 0.24 (p≤0.05) | | 0.18(ns) | |
The findings in this study support the earlier evidence that the dose-response effect of ondansetron in reduction of alcohol consumption is not linear. Of the doses used in this study, only 4 µg/kg (0.28 mg for a 70 kg person) bid exhibited clinically and statistically meaningful improvements in all efficacy endpoints. This study also suggested that ondansetron may be an appropriate therapy for EOA, but not LOA.
An Open-Label, 8-Week Study Comparing Ondansetron Effect in Early-Onset and Late-Onset Alcoholic Subjects
In 2003, Kranzler et al. reported on the co-administration of weekly cognitive behavioral therapy and ondansetron at 4 µg/kg bid for 8 weeks to 40 alcohol-dependent subjects (Kranzler, HR et al., 2003). The subjects were evenly divided between early-onset alcoholism (EOA; age of onset of the disorder <25 years) and late-onset alcoholism (LOA; age of onset of the disorder ≥25 years). Endpoints included drinks per day, DDD, PDA, Drinker Inventory of Consequences (DrInC) score, and percentage of heavy-drinking days, where heavy drinking was defined as ≥5 drinks in a day for a male subject or ≥4 drinks in a day for a female subject. Analyses were conducted comparing pre-treatment with 8-week values within onset category (EOA or LOA) and comparing treatment effects between categories.
The table below sets forth treatment results. All efficacy parameters improved significantly on treatment in both groups. EOA subjects reported significantly greater improvements in drinks per day, DDD, and DrInC score than LOA subjects. These findings, as noted earlier by Johnson et al., suggest that ondansetron shows promise for treatment of EOA by improving drinking outcomes.
Results of Study Comparing Effects of Ondansetron in EOA versus LOA
| | | EOA | | | LOA | | EOA v
LOA |
| | | change
mean
(SD) | | | p-value | | | change
mean
(SD) | | p-value | | | p-value |
| Drinks/drinking day | | | 5.78 (8.9) | | | | 0.009 | | | 1.55 (2.0) | | | 0.004 | | | | 0.032 |
| Drinks/day | | | 4.53 (4.5) | | | | <0.001 | | | 1.98 (2.1) | | | 0.001 | | | | 0.013 |
| Days abstinent (%) | | | 30.2 (29.4) | | | | <0.001 | | | 24.8 (21.2) | | | <0.001 | | | | 0.373 |
| Heavy-drinking days (%) | | | 35.1 (24.7) | | | | <0.001 | | | 26.7 (27.4) | | | <0.001 | | | | 0.139 |
| DrInC total score | | | 30.3 (27.7) | | | | <0.001 | | | 11.4 (11.2) | | | <0.001 | | | | 0.013 |
A 3-Period Study of Ondansetron Effect and Sertraline Effect in Subgroups of Alcoholics Constructed Based on Genotypes of the Serotonin Transporter Gene
Constructed Based on Genotypes of the Serotonin Transporter Gene
In 2009, Kenna et al. reported on a placebo-controlled cross-over study in which 21 alcohol-dependent subjects received 0.5 mg/day ondansetron or 200 mg/day sertraline for 3 weeks, placebo for 3 weeks and the alternative active medication for 3 weeks (Kenna, GA et al., 2009). An alcohol self-administration experiment was conducted at the end of each treatment period. The primary endpoint was DDD during the final week of each treatment period.
During the first 3-week treatment period, ondansetron-treated subjects carrying L/L genotype (n = 3), compared to the L/S and S/S carriers (n = 4), had a significantly fewer DDD (3.66 vs. 8.40, p = 0.02). Within L/S and S/S group, there was no significant effect of ondansetron. A pronounced order effect confounded analyses after the third 3-week treatment period.
Our clinical development program is designed to demonstrate the safety and efficacy of ondansetron in the alcohol-dependent population in low dosages for long periods of time, while targeting genotypes that have been shown to benefit from ondansetron treatment. Ultimately, this development program aims to establish a scientific link between the biology of alcohol addiction and the therapeutic mechanism of ondansetron action, permitting genetically-based prediction of ondansetron effectiveness.
Phase 2b Investigator Initiated Clinical Trial of AD04 for Alcohol Use Disorder Conducted by the University of Virginia
In various studies, it has been shown that alcohol dependent individuals with the LL genotype of the 5’-HTT and the TT genotype in the 3’-UTR LL and TT genotype have lower B-CIT neuronal binding to 5-HTT. It is hypothesized that individuals with the LL or TT genotype, 5-HTT gene expression is suppressed by increased alcohol consumption, and therefore, ondansetron, which causes 5-HTT gene expression would have the greatest effect upon individuals that possess both the LL genotype of the 5’-HTT and the TT genotype in the 3’-UTR. A subsequent Phase 2b study (N = 283), conducted by the University of Virginia for which we have acquired rights to the data, showed that a prospectively identified subgroup of alcohol-dependent individuals with these specific polymorphisms of the serotonin transporter protein responded therapeutically to ondansetron administration (Johnson, BA et al., 2011). Further analysis of this same data set against 18 additional polymorphisms located on the genes for the A and B subunits of the serotonin 5-HT3 receptor revealed polymorphisms that were also associated with a therapeutic response to ondansetron. Collectively, the genotypes from the two aforementioned analyses comprise the genotypes selected for testing in Phase 3 trials for AD04. The current ONWARD Phase 3 study is testing ondansetron’s efficacy compared with placebo based on its ability to decrease the frequency and amount of heavy drinking among alcohol dependent individuals with the selected genotypes.
Phase 2b Clinical Trial Study Design
The Phase 2b clinical trial conducted by the University of Virginia was a 283-patient, 12-week, randomized, two-center, parallel-group, placebo-controlled study. Following a 1 week placebo run in (single-blind), alcohol-dependent subjects were randomized to receive either 4 µg/kg ondansetron or placebo, orally, twice daily (double-blind) for 11 additional weeks. In addition to study treatment, all subjects received weekly, standardized, manual-driven, cognitive behavioral therapy.
Eligible subjects were classified to one of twelve groups described by the 2×2 x 3 factorial combinations and randomized to placebo or ondansetron (4 mcg/kg twice daily [b.i.d.]) using a computed blocks randomization procedure that balances the twelve treatment groups on drinks/day ≤ 7.99 vs ≥8.00), age of onset (early vs. late), and genotype (LL, SS, SL).
Genotyping and analysis of the study subjects for the SNP rs1042173 (TT, TG or GG) in the 3´-UTR of the 5-SLC6A4 gene that codes for the serotonin transporter was performed following randomization but prior to database lock. Genotyping and analysis of the study subjects for SNPs located on genes that govern expression of the 5-HT3A and 5-HT3B subunits of the 5-HT3 receptor was performed after database lock.
During treatment, subjects were evaluated weekly at the study center for efficacy, safety, and tolerability. Alcohol consumption was collected via the self-reported Timeline Follow-Back (TLFB) method (Sobell and Sobell, Psychosocial & Biochem. Meth., 1992).
Efficacy measures were based on self-reported drinking outcomes with drinks per drinking day (“DDD”), with a standard drink equal to 14 grams of alcohol, and the percentage of days abstinent (“PDA”) being the pre-specified efficacy end points. Withdrawal symptoms, social functioning, and motivation to use alcohol were assessed using standard questionnaires and scales. Subject safety was monitored through periodic electrocardiograms (EKGs), physical exams, safety laboratories and collection of adverse events, concomitant medications, and vital signs. Additionally, a post hoc analysis was conducted using the endpoint of percentage of heavy drinking days (“PDHD”), which is the number of days of heavy drinking days in a month as a percentage of days in the month, because it is widely recognized as a clinically meaningful endpoint and is expected to be an end point in a pivotal/Phase 3 trials. The PDHD end point requires that each day be determined to be a heavy drinking day (i.e., a day in which a female drinks 4 or more drinks or a male drinks 5 or more drinks) or not, making each day binary and requiring an increased sample size to ensure statistical power. Therefore, the goal of the PDHD analysis was to determine if the was a trend toward and effect with PDHD without necessary achieving statistical significance.
The study objectives were to evaluate the safety of AD04 and to test the hypotheses that: (i) ondansetron will have a greater effect of reducing the severity of alcohol drinking and of increasing the percentage of days abstinent among alcohol-dependent subjects with the LL genotype as compared with S carriers (SS or SL) of the 5´-HTTLPR; and (ii) ondansetron’s therapeutic effect will be greatest among alcohol-dependent subjects who possess both the LL genotype of the 5´-HTTLPR and the TT genotype of rs1042173 in the 3´-UTR of the 5´-HTT. After completion of the study, a planned additional analysis of the correlation between genotype and drinking outcomes was conducted considering 18 SNPs located on the 5-HT3A and 5-HT3B subunit genes that were selected based on their minor allele frequency (≥ 0.05) in different ethnic populations, to obtain uniform physical coverage of the two genes, and on results from previous genetic association studies. This latter analysis identified three SNPs as having an apparent beneficial effect.
The primary analytic procedure used mixed-effects linear regression models and a sensitivity analysis using repeated measures models.
Additionally, based on the expectation that subjects with the LL and LL/TT variants of the SLC6A4 gene would respond to ondansetron treatment while others do not, the possibility that SNPs in the 5-HT3A and 5-HT3B subunits of the 5-HT3AB receptor complex may also influence the response to ondansetron was planned as a post hoc analysis. The possible role of SNPs on the HTR3A and HTR3B genes in the response to ondansetron is logical since the 5-HT3A receptor subunit is the primary target for ondansetron’s actions, and the 5-HT3B receptor subunit may be associated with the availability and externalization of the 5-HT3AB receptor complex. Thus, alterations in post-synaptic receptors, such as the 5-HT3AB receptor complex, could have a large impact on signal transduction along post-synaptic neurons. For these analyses, a total of 18 SNPs on the genes for the 5-HT3A and 5-HT3B subunits were examined. SNPs were selected based on their minor allele frequency (≥ 0.05) in different ethnic populations, to obtain uniform physical coverage of the two genes, and on results from previous genetic association studies.
Summary Results — Safety:
Overall, 95% of the subjects in the ondansetron group and 96% in the placebo group reported a treatment-emergent AE (TEAE) during the study. TEAEs occurred most frequently in the SOCs of gastrointestinal disorders (ondansetron 65%, placebo 61%), metabolism and nutritional disorders (38%, 43%), and nervous system disorders (60%, 58%). The incidence of TEAEs by preferred term was similar between the ondansetron and placebo groups. TEAEs that occurred at a frequency ≥ 5% in the ondansetron group compared with the placebo group included constipation (32%, 21%), fatigue (39%, 25%), and dizziness (21%, 12%). There was one death during the study; Subject #218 committed suicide on Study Day 40. The event was considered not related to study drug. Treatment-emergent SAEs were reported in 3 (2.1%) ondansetron-treated subjects and 6 (3.8%) placebo-treated subjects. No SAE was considered related to study drug, and detoxification was the only SAE that was reported for more than 1 subject (2 ondansetron subjects). No clinically meaningful changes in clinical laboratory results, vital sign measurements, ECGs or physical examinations were observed for subjects during the course of the study.
Summary Results Phase 2b Clinical Trial — Primary Analysis of Efficacy of LL and LL/TT
Analysis of the LL genotype of the 5´-HTTLPR as compared to the non-LL genotypes showed a significant reduction in DDD and PDA (Johnson, et.al, Am. Jrnl. Psych., 2011). However, the demonstrated effect of the LL/TT vs. other patients was more pronounced, and carriers of LL/TT genotype who received ondansetron showed a greater reduction in drinking compared to LL/TT on placebo. Carriers of the LL/TT genotype who received ondansetron showed a greater reduction in DDD compared to: 1) LL/TT carriers who received placebo (difference of 2.05 drinks/drinking day; 95% CI, -3.72 to -0.39; p=0.0158), 2) LL/Gx carriers who received ondansetron (difference of 2.29 drinks/drinking day; 95% CI, -3.99 to -0.72; p=0.0048), and 3) all other genotypes who received ondansetron treatment (difference of 2.58 drinks/drinking day; 95% CI, -3.94 to 1.22; p<0.0001); and a greater PDA compared with: 1) the LL/TT genotype group treated with placebo (mean difference=12.38%; 95% CI= -1.57 to 26.33; p= 0.0819), 2) LL/Gx carriers treated with ondansetron (mean difference=15.14%; 95% CI= 1.41 to 28.87; p= 0.0307), and 3) all other genotypes treated with ondansetron (difference= 16.82%; 95% CI= 6.15 to 27.48; p=0.0020). The post hoc analysis of the PDHD endpoint show that ondansetron treatment of subjects with the LL/TT genotype was associated with a larger (but not statistically significant) reduction in PDHD compared to changes in PDHD in subjects with all other genotypes who received treatment with ondansetron (mean difference= -8.49%; 95% CI= 20.34 to 3.367; p= 0.1601). Similar trends (i.e., augmented reductions in PDHD) were observed for the LL/TT group treated with ondansetron versus the LL/Gx genotype group treated with ondansetron and versus the LL/TT group treated with placebo (mean difference=-2.54% 95% CI= 17.74 to 12.66, p=0.7431; and mean difference= 5.72% 95% CI= 21.20 to 9.75, p=0.4684; respectively).
Identification of Modulators of the 5-HT3 Receptor and Selection of the Phase 3 Genetic Panel for AD04
As stated above, a total of 18 SNPs on the genes for the 5-HT3A and 5-HT3B subunits were examined with SNPs selected based on frequency and on results from previous genetic association studies.
These analyses identified 3 SNPs (three in the gene for the 5-HT3A subunit and one in the gene for the 5-HT3B subunit) that were significantly associated with a positive response to ondansetron based on reductions in DDD and PDA. Thus, the genotype profile targeted for Phase 3 development is defined as those subjects who carry the LL/TT genotype and/or one of three 5-HT3 SNPs of interest (i.e., rs1150226-AG and rs1176713-GG in the gene that encodes the 5-HT3A receptor subunit and rs17614942-AC in the gene that encodes the 5-HT3B receptor subunit). The hypothesis that subjects who are carriers of the genotype panel targeted for study in Phase 3 (“P3-genotype”, with such patients “genotype positive” or “marker positive”) preferentially respond to treatment with ondansetron compared to subjects who do not carry any of the genotypes targeted for study in Phase 3 were assessed using the drinking endpoints of DDD, PDA, and PDHD.
Carriers of the P3-genotype who received ondansetron showed a greater reduction in DDD compared to P3-genotype carriers who received placebo (difference of 1.71 drinks/drinking day; 95% CI= -2.88 to -0.54; p=0.0042), and compared to subjects treated with ondansetron who were not carriers of the P3-genotype (All Other-OND; difference of 2.05 drinks/drinking day; 95% CI= -3.11 to -1.00, p=0.0001). In contrast, no difference was observed between non-P3-genotypes who received ondansetron (All Other-OND) versus non-P3-genotypes who received placebo (All Other-Placebo; difference of 0.40 drinks/drinking day; 95% CI= -0.43 to 1.23; p=0.3445). The mean baseline DDD for all subjects was 9.5 drinks/drinking day. Carriers of the P3-genotype who received ondansetron (P3-OND) had a greater increase in PDA compared to P3-genotype carriers who received placebo (P3-Placebo; difference of 11.56%; 95% CI= 0.80 to 22.31; p=0.0352) and compared to non-P3-genotype carriers who received ondansetron (All Other-OND; difference of 11.52%; 95% CI= 1.76 to 21.28; p=0.0208). In contrast, no differences were observed for the PDA endpoint between non-P3-genotypes treated with ondansetron versus non P3-genotypes treated with placebo (All Other-OND versus All Other-Placebo; difference of -0.96%; 95% CI= -8.61 to 6.69; p=0.8055). The mean baseline PDA for all subjects was 17%.
The results are summarized in the below graphs.
Phase 2b Clinical Trial Results — Analysis of Primary and Secondary Efficacy Endpoints for Target Genotypes
A 12-week, randomized, two-center, parallel-group, double-blind, placebo-controlled, two-arm (four cell) clinical trial of oral ondansetron (n=283)
As stated, above, the study was not powered to achieve statistical significance against the binary-by-day end point of PDHD, however, carriers of the P3-genotype who received ondansetron (P3-OND) showed a significantly greater reduction in PDHD compared to P3-genotype carriers who received placebo (P3-Placebo; difference of -11.08%; 95% CI= -21.90 to 0.27; p=0.0445), and compared to non-P3-genotype carriers who received ondansetron (All Other-OND; difference of -10.35%; 95% CI= -20.11 to -0.58; p=0.0378). In contrast, no difference was observed between non-P3-genotypes who received ondansetron (All Other-OND) versus non-P3-genotypes who received Placebo (All Other-Placebo; difference of 2.88%; 95% CI= -4.80 to 10.56; p=0.4625). The mean baseline PDHD for all subjects was 70%.
The results are summarized in the below graphs.
Phase 2b Clinical Trial Results — Post Hoc Analysis of Effect on Percentage of Heavy Drinking Days (defined as 4/5 or more drinks in a day for a woman/man, respectively)
A 12-week, randomized, two-center, parallel-group, double-blind, placebo-controlled, two-arm (four cell) clinical trial of oral ondansetron (n=283)
Definition of Heavy Drinking Day
As stated above, for the PDHD post hoc analysis of the Phase 2b clinical trial data, a heavy drinking day was defined as a day when a female drank 4 or more drinks in a day, with a drink being defined as containing 14 grams of alcohol, or when a man drank 5 or more drinks in a day, which was the definition the FDA indicated to us was required. It is also currently the definition of “high-risk drinking” in Dietary Guidelines for Americans 2015-2020 (U.S. Departments of HHS and Agriculture), the NIAAA’s definition of “binge drinking”, and has historically been the definition for a heavy drinking day (Neal, D., & Carey, K., 2007). The Substance Abuse and Mental Health Services Administration (SAMHSA) defines heavy drinking “as drinking 5 or more alcoholic drinks on the same occasion.” Subsequent to our analysis of the Phase 2b data and agreement with the FDA on the definition of a heavy drinking day as 4/5 or more drinks in a day for females/males, the FDA published a draft guidance, in which it states, “Those drinking 4 plus/5 plus [drinks for females and males, respectively] even on occasion have significantly higher risks (10 to 20 percent) of meeting criteria for AUD.” The FDA’s draft guidance then states that the NIAAA defines a heavy drinking day as more than 3 drinks in a day for a woman and more than 4 drinks in a day for a man, which is currently only part of the NIAAA’s definition for “low-risk drinking”, and which is very similar but not necessarily identical to what the FDA indicated to us was required and the criteria we used when generating our study report on the Phase 2b. So, it is unclear which definition of a heavy drinking day the FDA will accept at this time. However, under this different definition of a heavy drinking day as more than 3/4 for females/males, the Phase 2b trial data support the effect of AD04 on reducing heavy drinking and showed a greater reduction in PDHD compared to P3-genotype carriers who received placebo (P3-Placebo; difference of -10.24%; 95% CI= -21.18 to 0.70; p=0.0665), and compared to non-P3-genotype carriers who received ondansetron (All Other-OND; difference of -11.65%; 95% CI= -21.54 to -1.77; p=0.0209). In contrast, no difference was observed between non-P3-genotypes who received ondansetron (All Other-OND) versus non-P3-genotypes who received Placebo (All Other-Placebo; difference of 4.09%; 95% CI= -3.70 to 11.88; p=0.3033). We do not expect a small change to the definition of a heavy drinking day to dramatically change our plans or probability of success. We intend to discuss the definition of a heavy drinking day with the FDA and EMA prior to our relevant submissions.
Ongoing Phase 3 Clinical Program
The FDA has indicated that we can proceed with a single-arm, two-cell Phase 3 clinical trial design for the testing of AD04 as a treatment for AUD in patients that are genotype positive when tested against the AD04 genetic panel using our companion diagnostic test (i.e., a negative genetic test result will be an exclusion criterion). The initial Phase 3 trial is planned to be conducted in 290 patients in Scandinavia and Central and Eastern Europe where the prevalence of genotype positive people appears to be higher than in the U.S. and Western Europe. In January 2020 we announced that we had received favorable opinions from the Finnish Medicines Agency (FIMEA) and National Committee on Medical Research Ethics (TUKIJA) to commence our ONWARD pivotal Phase 3 clinical trial to investigate AD04 as a genetically targeted therapeutic agent for the treatment of AUD, and initiated the ONWARD trial in February 2020. As of December 31, 2020, 25 clinical sites across the countries of Sweden, Finland, Poland, Latvia, Estonia, Bulgaria and Croatia were actively enrolling patients, and the Phase 3 clinical trial was more than 50% enrolled. Data readout is currently expected by the first quarter of 2022.
The primary analysis is expected to use the primary endpoints previously accepted by the European Medicines Authority (“EMA”) with the reduction from baseline of heavy drinking and reduction from baseline in total alcohol consumed being the co-primary endpoints, and an alternative analysis is expected to be conducted for filing in the United States using the FDA specified endpoint of reduction in percentage of patients with heavy drinking during the efficacy observation period as compared to placebo (FDA Feb. 2015 Draft Guidance Alcoholism: Developing Drugs for Treatment Guidance for Industry ) and which the FDA has indicated will be acceptable. Under this guidance, the FDA appears to now define a heavy drinking as more than three drinks in a day for a woman and more than four drinks in a day for a man, which is a reduction from the prior definition. We intend to seek clarification from the FDA on the definition of a heavy drinking day prior to our submission to them and do not believe a minor change to the definition of a heavy drinking day will be material to our plans.
If the ONWARD trial is successful, we intend to consult with the FDA and EMA, and assuming agreement from the agencies, conduct a second Phase 3 clinical trial in a broader geography that includes the United States. The trial design is expected to be the same as ONWARD trial but is expected to include 580 patients in order provide increased exposure data to demonstrate the safety and tolerability of AD04 and increase the statistical power of the study. Depending on the results of the ONWARD trial, which is be fully powered for the FDA endpoint, it is also possible that the FDA may require a third Phase 3 trial. If a third Phase 3 trial is required, we would expect to conduct it in parallel with the second Phase 3 trial with a goal of not delaying approval of AD04.
We have had a joint meeting with the Center for Drug Evaluation and Review (“CDER”) and the Center for Devices and Radiological Health (“CDRH”), the two divisions of the FDA responsible for drug approvals and test approvals, respectively. At the meeting the divisions agreed that clinical validation of our companion diagnostic test for AD04 will be evaluated by CDER and the technical validation of our companion diagnostic will be evaluated by CDRH. We already developed the methods for the companion diagnostic as a blood test and established the test with a third-party vendor capable of supporting a Phase 3 clinical trial, and have built validation and possible approval of the companion diagnostic into the Phase 3 program, including that we plan to store blood samples for all patients in the event additional genetic testing is required by regulatory authorities.
We plan to test AD04 in adolescent patients (ages 12-17) as part of our next Phase 3 trial. If successful, we intend to request labeling for treating adolescent patients.
In parallel with the second Phase 3 trial, we expect to conduct any standard Phase 1 studies required by the regulatory agencies. Studies that have been discussed with the FDA as potentially being required might assess food effects, potentiation of the central nervous system effects of alcohol, and pharmacodynamic impact of certain cytochrome P450 enzyme variants.
License with University of Virginia Patent Foundation
In January 2011, we entered into an exclusive, worldwide license agreement with UVA LVG for rights to make, use or sell licensed products in the United States based upon the patents and patent applications made and held by UVA LVG (the “UVA LVG License”). Three patent and patent application families are included in the UVA LVG License, with patents issued in over 40 countries, including, without limitation, in the U.S., Europe and Eurasia. The licensed patents and patent applications currently include the below listed U.S. patents and patent application and any divisional patents, continuation patents and foreign equivalents.
| 1. | U.S. Patent Number 8,697,361, filed 1/11/11 |
“Serotonin Transporter Gene and Treatment of Alcoholism”
| 2. | U.S. Patent Number 8,753,815, filed 8/20/12 |
“Molecular genetic approach to treatment and diagnosis of alcohol and drug dependence”
| 3. | U.S. Patent Number 9,539,242, filed 4/30/14 |
“Molecular genetic approach to treatment and diagnosis of alcohol and drug dependence”
| 4. | U.S. Patent application number 15/848,079, filed 12/20/2017 |
“Molecular genetic approach to treatment and diagnosis of alcohol and drug dependence”
Additionally, the UVA LVG License grants rights to data and know-how developed by the University of Virginia related to AD04, including, without limitation, to the data from the Phase 2b study described above.
As consideration for the rights granted in the license agreement, we are obligated to pay UVA LVG yearly license fees and milestone payments, and a royalty based on net sales of products covered by the patent-related rights set forth above. More specifically, upon commencement of the license we issued to UVA LVG Class A Units (which was equal to four percent (4%) of our equity on the date of issuance) as a license issue. We are obligated to pay UVA LVG (i) annual minimum royalties of $40,000 commencing in 2017; (ii)a $20,000 milestone payments that as originally due upon dosing the first patient under a Phase 3 human clinical trial of a licensed product but has been paid in full, $155,000 upon the earlier of the completion of a Phase 3 trial of a licensed product or the partnering of the licensed or sale of our company, $275,000 upon acceptance of an NDA by the FDA, and $1,000,000 upon approval for sale of AD04 in the U.S., Europe or Japan; and (iii) royalties equal to a 2% and 1% of net sales of licensed products in countries in which a valid patent exists or does not exist, respectively, with royalties paid quarterly. In the event of a sublicense to a third party, we are obligated to pay royalties to UVA LVG equal to a percentage of what we would have been required to pay to UVA LVG had we sold the products under sublicense ourselves. In addition, we are required to pay to UVA LVG 15% of any sublicensing income. The license agreement, as amended on December 14, 2017 and further amended on December 18, 2019 and December 31, 2019 sets forth specific milestones completion deadlines including using commercially reasonable efforts to submit an NDA by December 31, 2024 and commence commercialization of an FDA approved product by December 31, 2025. The license agreement may be terminated by UVA LVG upon sixty (60) days written notice if we breach our obligations thereunder, including failing to make any milestone, or failing to use commercially reasonable efforts to submit an NDA or commence commercialization within the date specified above, failing to make other required payments, or the failure to exercise diligence to bring licensed products to market. In the event of a termination, we will be obligated to pay all amounts that accrued prior to such termination. The license agreement also contains other customary clauses and terms as are common in similar agreements between industry and academia, including agreements to indemnify UVA LVG for any liabilities arising out of or related to the licensee’s exercise of its rights under the license agreement, making the license grant subject to the Bayh-Dole Act (35 U.S.C. 200 et seq.), the reservation of the licensor of the right to use the licensed intellectual property rights for its internal, non-commercial purposes, limitations/disclaimers of various warranties and representations, reporting and record-keeping requirements, and licensee liability insurance requirements.
The term of the license continues until the expiration, abandonment or invalidation of the licensed patents, and following any such expiration, abandonment or invalidation will continue in perpetuity on a royalty-free, fully paid basis.
The UVA LVG currently has a policy under which up to 35% of the payments made to the UVA LVG under a license may be distributed to inventor of the licensed technology, therefor the Chairman of the Board in his capacity as inventor of the patents licensed by us from the UVA LVG may be eligible to receive such payments from the UVA LVG.
PEPCO MSA
On July 5, 2019, we entered into a Master Services Agreement (the “MSA”) and attached statement of work (the “SOW”) with Psychological Education Publishing Company (“PEPCO”) to administer a behavioral therapy program during our upcoming Phase 3 clinical trial using AD04, for the treatment of alcohol use disorder. Specifically, PEPCO is engaged in the business of training and certifying clinical investigators in the administration of Brief Behavioral Compliance Enhancement Treatment (“BBCET”). PEPCO is owned by Dr. Bankole Johnson, our Chief Medical Officer, and currently our largest stockholder. We may terminate the MSA at any time upon ten (10) days prior written notice to PEPCO. Unless otherwise indicated in our notice of termination, Work (as defined in the MSA) under any statement of work in progress at the time of the delivery of notice of termination shall continue as if the applicable statement of work had not been terminated, and the terms hereof shall continue to apply to such work. We may also terminate the MSA for cause due to PEPCO’s failure to perform its obligations thereunder upon three (3) days prior written notice to PEPCO; provided, however, the Company may terminate the MSA immediately in the event of PEPCO’s violation, or threatened violation, of certain provisions contained therein.
The statement of work under the MSA will terminate upon the completion the final study report for the Trial and delivery of the final report by PEPCO on the supervision and monitoring of the BBCET, including, without limitation, data reports. Notwithstanding the forgoing, the statement of work may be terminated by us upon written notice to PEPCO.
Prior to amendment (see below), it was anticipated that the compensation to be paid to PEPCO for services under the MSA would be approximately $300,000, of which subject to approval of the Nasdaq Capital Market shares of our common stock having a value equal to twenty percent (20%) of the fees due thereunder (the “Company Shares”) would be issued to Dr. Johnson as a consultant under the 2017 Equity Incentive Plan. On October 2, 2019, the Company issued 3,187 shares of common stock to Dr. Johnson at a market price of $1.51 per share and total value of $4,812 under the terms of the MSA.
On December 12, 2019, we entered into an Amendment (the “Amendment”) to the SOW. We had paid PEPCO $39,064 under the SOW for services rendered to date, leaving as estimated balance of $274,779 estimated to be paid under the SOW. The Amendment provided us with a 20% discount on the remaining services to be provided under the SOW and fixed the price of any remaining services under the SOW to be a total of $219,823 for all services required for the use of Brief Behavioral Compliance Enhancement Treatment (BBCET) in support of Phase 3 clinical trial provided that payment be made no later than December 13, 2019, which payment was made. As of December 31, 2020, the Company had recognized $147,120 in expenses associated with this MSA, of which $108,056 were charged against cash advanced under the terms of the Amendment, leaving a net prepaid expense asset of $111,767.
In addition, Dr. Johnson executed a guaranty, dated December 12, 2019, of PEPCO’s performance under the MSA and SOW (the “Guaranty”), together with a pledge and security agreement, dated December 12, 2019 (the “Pledge and Security Agreement”), to secure the Guaranty with 600,000 shares of our common stock beneficially owned by him and a lock-up agreement, dated December 12, 2019 (the “Lock-Up”), pursuant to which he agreed not to transfer or dispose of, directly or indirectly, any shares of our common stock, as currently owned by him, until after January 1, 2021.
On August 19, 2020, we and Dr. Bankole Johnson entered into a Lock-Up Agreement Extension and Right of First Refusal (the “Lock-Up Extension”), which amended the Lock-Up Agreement that they had entered into dated December 12, 2019 (the “Lock-Up”). The Lock-Up Extension extended the term of Dr. Johnson’s Lock-Up from January 1, 2021 until April 1, 2021. In connection with the Lock-Up Extension, Dr. Johnson was released from his Lock-Up restrictions with respect to 350,000 shares of our common stock, in order to enable Dr. Johnson to fund his new clinic focused on brain wellness and addiction treatments, Privée Clinics, LLC. Additionally, under the Lock-Up Extension, we were granted a right of first refusal for future financings in Privée Clinics, LLC
Protection from Generic Competition
Since our inception, we have focused on taking action primarily through the filing of patents geared toward ensuring AD04 will have market exclusivity for at least 10 years after it is launched with particular focus on the U.S. and Europe. Ondansetron, the active pharmaceutical ingredient (“API”) of AD04 was granted FDA approval as Zofran ® for the treatment of post-operative and post-chemotherapy nausea and emesis in January 1991 and is now commercially available in generic form at doses from more than 12 times the AD04 dose to over 70 times the AD04 dose with the highest doses being administered intravenously (“i.v.”), which provides almost twice the drug exposure levels as oral dosing. With generic ondansetron available, the following threats have been addressed: (i) the potential use of currently available ondansetron products (i.e., Zofran ®) “off-label”, and (ii) the potential manufacturing and launching of a generic version AD04 by a competitor.
Limited Threat of “Off-label” Use of Zofran ®
The lowest doses of Zofran ® tablets (and its generic equivalents) on the market are a 4 mg and 8 mg tablet as compared to AD04, which is currently formulated as a 0.33 mg tablet (12.2 times less than the 4 mg tablet). Thus, in order for a patient to use tablets already on the market and get the AD04 dose, a patient would have to cut the 4 mg tablet into 12 parts (or the 8 mg tablet into 24 parts), which we do not believe is reasonably possible; and, even with precise sectioning into 12 pieces, the dose may still not be accurate because tablets at the Zofran ® dose have not been manufactured to ensure uniformity of distribution of the active ingredient across the tablet. Therefore, we believe that the risk of a large number of patients attempting to cut the currently marketed tablet to achieve the AD04 dose to be extremely low.
Since we do not believe that Zofran ® tablets can be used as a substitute for AD04, the main question related to the potential for off-label use of the current products for treating addictions then becomes whether doctors and patients will believe it is possible to use the currently available, higher doses of ondansetron to treat addictions, including AUD. We believe doctors are extremely unlikely to prescribe currently available high dose versions of ondansetron and that any such prescribing that dose will likely be limited and immaterial to the sales of AD04 for two reasons — (1) we believe the high doses are unlikely to be efficacious as a treatment for AUD, and (2) we believe the high doses would likely raise significant safety concerns.
| 1. | Lack of Efficacy. The high doses of ondansetron found in Zofran ® have been tested in clinical trials for treating AUD and have not shown efficacy against AUD (Sellers, et. al. 1994). At best, existing trial results do not suggest that the high Zofran®-level doses of ondansetron currently on the market and approved for nausea and emesis will be effective. |
| 2. | Safety Concerns. While high-dose ondansetron is safe and tolerable at the doses on the market if administered acutely (i.e., dosed for a few hours i.v. or a few days orally) as is done for post-operative and post-chemotherapy nausea and emesis, the drug is known to have cardiovascular side effects at higher doses, and results from clinical studies suggest that high doses of ondansetron may affect the electrical activity of the heart. In fact, the FDA withdrew approval of the 32 mg i.v. Zofran ® product that was previously on the market. As part of the FDA’s on-going safety review of currently available ondansetron doses, the FDA has stated that: “Ondansetron at currently marketed levels may increase the risk of developing prolongation of the QT interval of the electrocardiogram, which can lead to an abnormal or potentially fatal heart rhythm.” There are also several recent lawsuits claiming that Zofran ® used for off label for morning sickness causes birth defects. Thus, if the currently available high-dose ondansetron was used chronically as would be needed for treating addiction there could potentially be significant safety concerns without additional clinical studies related to the chronic dosing of currently available ondansetron. At the lower dose of ondansetron in AD04, our product is almost as low as one one-hundredth of the dose of i.v. ondansetron that was removed from the market. The FDA has stated that we can commence chronic dosing of patients with AD04 without any further safety or non-clinical studies. |
Therefore, we do not expect physicians to prescribe current ondansetron doses for currently unapproved use for treating AUD because there is no evidence those doses would work for treating AUD and there may be safety concerns associated with the chronic administration of currently available doses.
There is also a liquid, pediatric formulation of Zofran® on the market. It is offered in a 50 mL bottle that is available for a little over $100 online and would provide a 2-month supply of AD04 if dosed at the 0.4 mL required to achieve the 0.33 mg AD04 dose. Our risk assessment is that, though it would be possible to use the liquid formulation for administering a dose of ondansetron equivalent to AD04, it is not expected to be a practice that would materially impact the sales of AD04, and the risk from the liquid formulation is low for the following reasons:
| 1. | Compliance concerns. In the field of addiction, patient compliance is one of the biggest concerns for both the physicians and the patients themselves. A treatment not appropriately administered is a treatment that will not work. Oral tablets have been shown to have one of the highest compliance rates over other dosage forms. It is likely that both physicians and patients will demand the tablet in order to improve compliance and, thus, treatment success rates. |
| 2. | Inconvenient, complicated delivery. A major driver of compliance is the convenience of appropriately administering the drug. Appropriate delivery of the liquid formulation would require patients to measure each dose into a graduated dropper or syringe (administration of such a small amount (0.4 mL) by graduated cup would not be practical). Cleanup of the sticky product would be inconvenient as would transportation and storage, and an opened bottle would need to be used within 4 weeks (per UKPAR). Therefore, we expect that AD04’s convenient tablet would increase patient compliance relative to the liquid formulation. Bottle breakage and spillage will also be a concern. |
| 3. | Dosing Accuracy. Dosing accuracy is particularly important when using ondansetron to treat alcoholism due to the limitations of the therapeutic window and the cardiovascular side effects at high doses. With the liquid formulation, measuring the small (0.4 mL) dose will be difficult with great opportunity for misdosing even if a graduated syringe is used. In real-world practice, many patients would use other methods such as estimated pouring into cups and drinking directly from the bottle. Misdosing could significantly affect the safety and/or efficacy of the treatment. |
| 4. | Lack of physician motivation to prescribe the liquid formulation. Given the known compliance advantages of oral tablets vs. liquid formulations, the heightened need for compliance in this particular patient population, and the concerns around dosing accuracy with a liquid formulation, we believe it is likely physicians would recognize the risk of prescribing the liquid formulation off-label and so be unwilling to prescribe it. For insured patients, any differential in co-payments would create little incentive to use the liquid formulation relative to the compliance and inconvenience problems. |
| 5. | Lack of competitive marketing. Manufacturers of liquid ondansetron are not allowed to market for reduction in alcohol use disorder because reduction in alcohol use disorder is not an approved indication for their product. Furthermore, most generic companies do not have marketing efforts of any kind. |
| 6. | Litigation risk to large prescribers. If a large clinic (such as a rehabilitation clinic) prescribes or provides the liquid formulation off-label, the institution could be liable for inducing infringement of our licensed patents. |
In summary, we do not expect off-label use of currently available ondansetron to meaningfully impact the sales of AD04.
Protection from a Competitor Launching a Generic Version of AD04.
We believe that we license the patent protection necessary to protect us against the launch by a competitor of a generic version of AD04. The label being sought for AD04 will be:
The use of AD04 (i.e., ondansetron) for the treatment of patients that are positive for the specified genetic markers.
The only use for the AD04 dose of ondansetron will be under this label.
Our licensed patents cover the following:
The use of AD04 (i.e., ondansetron) for the treatment of patients that are positive for the specified genetic markers.
We believe that any attempt by competitors to reformulate and market ondansetron at our intended dosage levels, while technically feasible, can be interpreted under current case law as inducement to infringe on our intellectual property rights, which should, accordingly, be actionable. Additionally, there will be no unpatented use for the AD04 dose of ondansetron. So, a competitor that sells a product containing the AD04 dose of ondansetron will indirectly infringe our licensed patents, which should, accordingly, be actionable.
A competitor could sell a dose equal to that of AD04 and avoid our licensed patents if they conduct a Phase 3 program using the AD04 dose to treat a different label indication and achieved successful results and approval. We do not know of any clinical development programs of ondansetron underway at this time and so consider this risk to be negligible.
Governmental Regulation
Our business is subject to extensive laws and regulations, the most significant of which are summarized below.
FDA Approval Process
In the United States, pharmaceutical products are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act (the “FDC Act”), and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. In the United States, pharmaceutical products used for the prevention, treatment, or cure of a disease or condition of a human being are subject to extensive regulation under the FDC Act. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Pharmaceutical product development for a new product or certain changes to an approved product in the United States typically involves preclinical laboratory and animal tests, the submission to the FDA of an investigational new drug application (“IND”), which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation, and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls, and a proposed clinical trial protocol. Long-term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin. However, the FDA can impose a clinical hold after 30 days if it has safety or compliance-related concerns.
Clinical trials involve the administration of the investigational new drug or biologic to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with federal regulations; (ii) in compliance with good clinical practice (“GCP”), an international standard meant to protect the rights and health of subjects and to define the roles of clinical trial sponsors, administrators, and monitors; as well as (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. subjects and subsequent protocol amendments must be submitted to the FDA as part of the IND.
As noted, the FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for subjects in clinical trials must also be submitted to an institutional review board (“IRB”), for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, for safety or other concerns, or may impose other conditions.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, the initial introduction of the drug or biologic into healthy human subjects or patients, the product is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence of effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug or biologic for a particular indication, dosage tolerance, and optimum dosage, and to identify common adverse effects and safety risks. If preliminary evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug or biologic and to provide adequate information for the labeling of the product. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug or biologic.
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the United States. The NDA must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture, and control. The cost of preparing and submitting an NDA is substantial. The submission of most NDAs is additionally subject to a substantial application user fee, currently exceeding $2.5 million for fiscal year 2019 (although a waiver is possible in certain cases), and the manufacturer and/or sponsor under an approved new drug application are also subject to a program fee set at more than $309,000 for fiscal year 2019. These fees are typically increased annually.
The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs. Most such applications for standard review drug or biologic products are reviewed within ten to twelve months; most applications for priority review drugs or biologics are reviewed in six to eight months. The FDA can extend these reviews by three months. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission.
The FDA may also refer applications for novel drug or biologic products, or drug or biologic products that present difficult questions of safety or efficacy, to an advisory committee — typically a panel that includes clinicians and other experts — for review, evaluation, and a recommendation on questions raised by an application, including whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with current good manufacturing practice (“cGMP”) is satisfactory and the NDA contains data that provide substantial evidence that the drug or biologic is safe and effective in the indication studied.
After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a Complete Response Letter (“CRL”). In some cases, FDA may choose to extend the review time, in consultation with the sponsor. A CRL generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included.
An approval letter authorizes commercial marketing of the drug or biologic with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use (“ETASU”). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the product. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the product’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing. The FDA could also impose a boxed warning (sometimes referred to as a Black Box Warning) in the product label if it identifies a specific risk that requires particular attention. This imposition of a Black Box Warning limits certain types of promotions.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented.
Enacted in 2016, the 21st Century Cures Act (the “Cures Act”), in part, revises the drug and device review and approval processes at the FDA. The Cures Act, which was signed into law on December 13, 2016, among other things, requires the manufacturer of an investigational drug for a serious disease or condition to make available, such as by posting on its website, its policy on evaluating and responding to requests for individual patient access to such investigational drug. This requirement applies on the later of 60 calendar days after the date of enactment of the Cures Act or the first initiation of a Phase 2 or Phase 3 trial of the investigational drug.
The FDA has various programs, including fast track designation, accelerated approval, priority review, and breakthrough therapy designation, which are intended to expedite or simplify the process for the development and FDA review of drugs that are intended for the treatment of serious or life-threatening diseases or conditions and demonstrate the potential to address unmet medical needs. We believe AD04 may qualify for one or more of these programs and intend to purse one or more of them as part of our strategy to expedite the approval of AD04 for marketing.
Post-Approval Requirements
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs and biologics, including standards and regulations for direct-to-consumer advertising, industry-sponsored scientific and educational activities and promotional activities involving the internet. Drugs and biologics may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA. The FDA also may require post-marketing testing, known as Phase 4 testing, REMS, and special surveillance to monitor the effects of an approved product, or the FDA may place other conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, drug manufacture, packaging, and labeling procedures must continue to conform to cGMPs after approval. Drug and biologic manufacturers must list the product with the FDA, and they and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing and other facilities to assess compliance with cGMPs and other requirements. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality-control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals, issue warning or other letters, suspend production activities, or request product recalls if a company fails to comply with regulatory standards, or take other regulatory or enforcement action if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered. Significant expenses are required to correct deficiencies.
Companion diagnostics and complementary diagnostics
We believe that the success of our product candidates may depend, in part, on the development and commercialization of either a companion diagnostic or complementary diagnostic. Companion diagnostics and complementary diagnostics can identify patients who are most likely to benefit from a particular therapeutic product; identify patients likely to be at increased risk for serious side effects as a result of treatment with a particular therapeutic product; or monitor response to treatment with a particular therapeutic product for the purpose of adjusting treatment to achieve improved safety or effectiveness. Companion diagnostics and complementary diagnostics are regulated as medical devices by the FDA and, as such, require either clearance or approval prior to commercialization. The level of risk combined with available controls to mitigate risk determines whether a companion diagnostic device requires Premarket Approval Application, or PMA, approval or is cleared through the 510(k) premarket notification process. For a novel therapeutic product for which a companion diagnostic device is essential for the safe and effective use of the product, the companion diagnostic device should be developed and approved or 510(k)-cleared contemporaneously with the therapeutic. The use of the companion diagnostic device will be stipulated in the labeling of the therapeutic product. This is also true for a complementary diagnostic, although it is not a prerequisite for receiving the therapeutic. Currently, we intend to submit a 505(b)(2) new drug application to the FDA for AD04. We have interacted primarily with the FDA’s Center for Drug Evaluation and Research, in consultation with the agency’s Center for Devices and Radiological Health. At this time, the FDA has not stated that a new marketing application (e.g., a PMA, or an approval cleared through the 510(k) premarket notification process) will be required for the companion diagnostics to be used with the drug product, but this could change. If the FDA requires a separate application for the diagnostic, this could potentially delay the approval of the new drug application for AD04, complicate the review process, or even lead to the rejection of the new drug application.
Hatch-Waxman Amendments to the Federal Food, Drug and Cosmetic Act
Under certain circumstances, an approved application may be eligible for three years of non-patent market exclusivity provided by the Hatch-Waxman Amendments to the Federal Food, Drug, and Cosmetic Act. The FDA might grant such exclusivity, (which would be separate from any patent protection to which an approved drug might be entitled) if the applicant conducted new clinical investigations (other than bioavailability studies) that are new and essential to the application’s approval. Among the types of exclusivity are those for a “new chemical entity” and those for a new formulation or indication for a previously-approved drug. If granted, marketing exclusivity for the types of products that include only drugs with innovative changes to previously-approved products using the same active ingredient, might prohibit the FDA from approving an application for a competitor product, such as an abbreviated new drug application or a 505(b)(2) NDA relying on the finding of safety and efficacy for three years. This three-year exclusivity, however, covers only the innovation associated with the original NDA. It does not prohibit the FDA from approving applications for drugs with the same active ingredient but without the new innovative change. These marketing exclusivity protections do not prohibit the FDA from approving a full NDA, even if it contains the innovative change. There is no guarantee that the FDA will grant such exclusivity and competitors can try to seek approval of competitive products, notwithstanding the exclusivity. However, if three years of exclusivity is afforded, it offers us one more barrier to competitor entry for a few years.
505(b)(2) NDA
We intend to submit a 505(b)(2) NDA. A 505(b)(2) NDA provided by Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act, allows the FDA to rely, for approval of an NDA, on data not developed by the applicant. Such an NDA, referred to as a 505(b)(2) application contains full reports of investigations of safety and effectiveness, but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Such applications permit approval of applications other than those for duplicate products and permit reliance for such approvals on scientific literature or an FDA finding of safety and/or effectiveness for a previously approved drug product. While each application is different, these types of applications will typically require bridging studies (to support the change or modification from the listed drug) and could require clinical data to support the modification of the already-approved drug product.
In addition, a 505(b)(2) NDA requires the applicant to certify as to any patents that claim the drug for which a claim of patent infringement could be made. In certain cases, the applicant of the NDA with a patent certification must provide notice to the patent holder, which can lead to a patent infringement lawsuit, thereby delaying the FDA approval of the competitor product for up to 30 months, separate from any traditional patent infringement litigation delay. Similarly, if the competitor has its own market exclusivity, this can delay approval of the product. However, if a product obtains exclusivity or patent protection, it can delay entry of competitors for several years.
Pediatric Information
Under the Pediatric Research Equity Act (“PREA”), NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant full or partial waivers, or deferrals, for submission of data. We plan to test AD04 in adolescent patients (ages 12-17) as part of our next Phase 3 trial. If successful, we intend to request labeling for treating adolescent patients.
Fraud and Abuse and Other Healthcare Regulation
We are subject to various federal and state healthcare laws, including, but not limited to, anti-kickback laws. Penalties for violations of these healthcare laws include, but are not limited to, criminal, civil and/or administrative penalties, damages, fines, disgorgement, individual imprisonment, possible exclusion from Medicare, Medicaid and other federal and state healthcare programs, and the curtailment or restructuring of operations.
Anti-Kickback Statute
The federal Anti-Kickback Statute prohibits persons or entities from knowingly and willfully soliciting, offering, receiving or paying any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, in exchange for or to induce either the referral of an individual, or the furnishing, arranging for or recommending a good or service, or for the purchasing, leasing, ordering, or arranging for or recommending, any good, facility, service or item for which payment may be made in whole or in part under federal healthcare programs, such as the Medicare and Medicaid programs. The federal Anti-Kickback Statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. The term “remuneration” expressly includes kickbacks, bribes, or rebates and also has been broadly interpreted to include anything of value, including for example, gifts, discounts, meals, entertainment, the furnishing of supplies or equipment, credit arrangements, payments of cash, waivers of payments, ownership interests and providing anything at less than its fair market value.
There are a number of statutory exceptions and regulatory safe harbors protecting certain business arrangements from prosecution under the federal Anti-Kickback Statute. These statutory exceptions and safe harbors set forth provisions that, if all their applicable requirements are met, will assure healthcare providers and other parties that they may not be prosecuted under the federal Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more applicable statutory exceptions or safe harbors does not necessarily mean that it is per se illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy all requirements of an applicable safe harbor may result in increased scrutiny by government enforcement authorities and will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Additionally, the intent standard under the federal Anti-Kickback Statute was amended under the Affordable Care Act, to a stricter standard such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. The Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act, which is discussed below.
Federal Civil False Claims Act
The federal civil False Claims Act prohibits, among other things, persons or entities from knowingly presenting or causing to be presented a false or fraudulent claim to, or the knowing use of false statements to obtain payment from or approval by, the federal government. Suits filed under the federal civil False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government. These individuals, sometimes known as “relators” or, more commonly, as “whistleblowers”, may share in any amounts paid by the entity to the government in fines or settlement. The number of filings of qui tam actions has increased significantly in recent years, causing more healthcare companies to have to defend a case brought under the federal civil False Claim Act. If an entity is determined to have violated the federal civil False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each separate false claim. Many comparable state laws are broader in scope and apply to all payors, and therefore, are not limited to only those claims submitted to the federal government.
Federal Physician Self-Referral Prohibition
We may also be subject to the federal physician self-referral prohibitions, commonly known as the Stark Law, which prohibits, among other things, physicians who have a financial relationship, including an investment, ownership or compensation relationship with an entity, from referring Medicare and Medicaid patients for designated health services (which include clinical laboratory services) to such entity, unless an exception applies. Similarly, entities may not bill Medicare, Medicaid or any other party for services furnished pursuant to a prohibited referral. Many states have their own self-referral laws as well, which in some cases apply to all third-party payors, not just Medicare and Medicaid.
Federal Civil Monetary Penalties Statute
The federal Civil Monetary Penalties Statute, among other things, imposes fines against any person or entity who is determined to have presented, or caused to be presented, claims to a federal healthcare program that the person knows, or should know, is for an item or service that was not provided as claimed or is false or fraudulent.
Health Insurance Portability and Accountability Act of 1996
The federal Health Insurance Portability and Accountability Act (“HIPAA”) created several new federal crimes, including healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payors. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services.
In addition, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), and their implementing regulations established uniform standards for certain covered entities, which are healthcare providers, health plans and healthcare clearinghouses, as well as their business associates, governing the conduct of specified electronic healthcare transactions and protecting the security and privacy of protected health information. Among other things, HITECH also created four new tiers of civil monetary penalties and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions.
The Federal Physician Payments Sunshine Act
The federal Physician Payment Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with certain exceptions, to report annually to CMS, information related to “payments or other transfers of value” provided to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and to report annually to CMS ownership and investment interests held by physicians, as defined above, and their immediate family members. Failure to submit timely, accurately and completely the required information for all payments, transfers of value and ownership or investment interests may result in civil monetary penalties of up to an aggregate of $150,000 per year and up to an aggregate of $1.0 million per year for “knowing failures.”
State Law Equivalents
Many states have also adopted laws similar to each of the above federal laws, such as anti-kickback and false claims laws, which may be broader in scope and apply to items or services reimbursed by any third-party payor, including commercial insurers, as well as laws that restrict our marketing activities with health care professionals and entities, and require us to track and report payments and other transfers of value, including consulting fees, provided to certain healthcare professionals and entities. Some states mandate implementation of compliance programs to ensure compliance with these laws. We also are subject to foreign fraud and abuse laws, which vary by country.
Healthcare Reform
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “ACA”), which has the potential to substantially change healthcare financing and delivery by both governmental and private insurers, and significantly impact the drug and medical device industries. The ACA will impact existing government healthcare programs and will result in the development of new programs.
In addition, the ACA and its implementing regulations, among other things, revised the methodology for calculation of rebates owed by manufacturers to the state and federal government for covered outpatient drugs and certain biologics, including AD04 or any future product candidates, under the Medicaid Drug Rebate Program, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations, subjected manufacturers to new annual fees and taxes for certain branded prescription drugs, and provided incentives to programs that increase the federal government’s comparative effectiveness research.
Other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers up to 2% per fiscal year. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012 (the “ATRA”) which delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. In March 2013, the President signed an executive order implementing sequestration, and in April 2013, the 2% Medicare payment reductions went into effect. The ATRA also, among other things, reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
In addition, Congress often uses the Medicare program for pay for legislation. For example, on April 16, 2015, President Obama signed into law the “Medicare Access and CHIP Reauthorization Act of 2015” (“MACRA”). MACRA repealed the Medicare sustainable growth rate formula that had been used to determine payment levels under the Medicare physician fee schedule (“PFS”), and established a new method to update payments for physicians and other providers paid under the PFS. Congress reduced Medicare payments for several categories of providers and made changes to Medicare policies to offset the cost of the bill. It is possible that future legislation and regulations may include Medicare payment reductions or policy changes that result in reduced payments, increased burdens or increased operating costs.
The full impact of the ACA, as well as other laws and reform measures that may be proposed and adopted in the future, remains uncertain, but may continue the downward pressure on medical device pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs, which could have a material adverse effect on our business operations. Efforts to significantly amend or repeal the ACA continue and if passed could have a significant impact on important aspects of our business including medical device and drug pricing, Medicare payment reductions or policy changes that result in reduced payments, or increased burdens or operating costs.
The Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act (“FCPA”), prohibits any U.S. individual or business from paying, offering, or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of such foreign official in her or her official capacity or to secure any other improper advantage in order to obtain or retain business. In addition to the antibribery provisions, the FCPA also obligates “issuers,” companies whose securities are registered pursuant to Section 12 of the Exchange Act or is required to file periodic and other reports with SEC under Section 15(d) of the Exchange Act to comply with the FCPA’s record keeping and internal controls provisions; the accounting provisions require a listed company to maintain books and records that, in reasonable detail, accurately and fairly reflect all transactions of the corporation, including international affiliates, and to devise and maintain an adequate system of internal accounting controls to assure management’s control authority, and responsibility over the company’s assets.
Export Controls and Economic Sanctions
Several U.S. statutes and regulations regulate the export from the United States of pharmaceutical products. Pursuant to the Export Administration Regulations, (“EAR”) the export (including re-exports and “deemed exports”) of commercial and “dual-use” products may require a license or be prohibited. A listing of the types of goods and services controlled for export by the EAR is on the Commerce Control List (“CCL”), which includes essentially all civilian science, technology, and engineering dual use items. For products listed on the CCL, a license will be required as a condition to export, unless an exclusion or license exception applies. Those items not explicitly included on the CCL are included in a broad category known as “EAR99.” Although a license may not generally be required for EAR99 designated items, a license will be required if the item will be shipped or otherwise transferred to a comprehensively embargoed country or for a potentially prohibited purpose.
The Commerce Department’s Office of Antiboycott Compliance and the Treasury Department’s Internal Revenue Service enforce anti-boycott compliance regulations that prohibit U.S. persons such as the Company from participating directly or indirectly with an economic boycott that is not recognized by the United States. The regulations include reporting requirements, prohibitions, and tax liabilities that may be incurred if the Company supports, even inadvertently, an economic boycott in which the U.S. does not participate.
Pursuant to the Trading With the Enemy Act, the International Emergency Economic Powers Act, and other related statutes, regulations, and Executive Orders, the Treasury Department’s Office of Foreign Assets Control (“OFAC”), administers and enforces economic and trade sanctions that prohibit or restrict certain activities with embargoed countries, sanctioned entities, and sanctioned individuals for particular foreign policy and national security reasons. The scope of the sanctions varies significantly, but may include comprehensive restrictions on imports, exports, investment, and facilitation of foreign transactions involving a sanctioned jurisdiction, entity or person, as well as non-sanctioned persons and entities acting on behalf of sanctioned jurisdictions, entities or people. OFAC’s programs also prohibit U.S. persons, such as the Company, from transacting with any person or entity that is deemed to be a Foreign Sanctions Evader (foreign individuals and entities determined to have violated, attempted to violate, conspired to violate, or caused a violation of U.S. sanctions).
Other U.S. government agencies, including the U.S. Department of State, may maintain regulations that impact the Company’s ability to export pharmaceutical products from the United States. These broad range of U.S. export control laws and regulations obligate U.S. businesses to develop, maintain, and enforce an adequate system of internal controls to ensure compliance with such laws and regulations.
Implications of Being an Emerging Growth Company
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), and therefore we intend to take advantage of certain exemptions from various public company reporting requirements, including not being required to have our internal controls over financial reporting audited by our independent registered public accounting firm pursuant to Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and any golden parachute payments. We may take advantage of these exemptions until we are no longer an “emerging growth company.” In addition, the JOBS Act provides that an “emerging growth company” can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have elected to use the extended transition period for complying with new or revised accounting standards under the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates. We will remain an “emerging growth company” until the earlier of (1) the last day of the fiscal year: (a) following the fifth anniversary of the completion of our initial public offering; (b) in which we have total annual gross revenue of at least $1.07 billion; or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeded $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period. References herein to “emerging growth company” have the meaning associated with that term in the JOBS Act.
Corporate Information
ADial Pharmaceuticals, L.L.C. was formed as a Virginia limited liability company in November 2010. ADial Pharmaceuticals, L.L.C. converted from a Virginia limited liability company into a Virginia corporation on October 3, 2017, and then reincorporated in Delaware on October 11, 2017 by merging the Virginia corporation with and into Adial Pharmaceuticals, Inc., a Delaware corporation that was incorporated on October 5, 2017 as a wholly owned subsidiary of the Virginia corporation. We refer to this as the corporate conversion/reincorporation. In connection with the corporate conversion/reincorporation, each unit of ADial Pharmaceuticals, L.L.C. was converted into shares of common stock of the Virginia corporation and then into shares of common stock of Adial Pharmaceuticals, Inc., the members of ADial Pharmaceuticals, L.L.C. became stockholders of Adial Pharmaceuticals, Inc. and Adial Pharmaceuticals, Inc. succeeded to the business of ADial Pharmaceuticals, L.L.C.
Our principal executive offices are located at 1180 Seminole Trail, Suite 495, Charlottesville, Virginia 22901, and our telephone number is (434) 422-9800. Our website address is www.adialpharma.com. Information contained in our website does not form part of this registration statement and is intended for informational purposes only.
This registration statement contains references to our trademarks and to trademarks belonging to other entities. Solely for convenience, trademarks and trade names referred to in this registration statement, including logos, artwork and other visual displays, may appear without the ® or TM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
Human Capital/ Employees
As of the date of this registration statement, we have nine employees, of which eight are full-time employees and one is a three-quarters time employee. Our Chief Medical Officer is a consultant that devotes 75% of his working time to providing services to us. None of our employees is represented by a labor union, and we consider our relationship with our employees to be good.
We believe our relationships with our employees are satisfactory. We anticipate that we will need to identify, attract, train and retain other highly skilled personnel to pursue our development program. Hiring for such personnel is competitive, and there can be no assurance that we will be able to retain our key employees or attract, assimilate or retain the qualified personnel necessary for the development of our business.
We have no collective bargaining agreements with our employees and have not experienced any work stoppages. We consider our relations with our employees to be good. Although, management continually seeks to add additional talent to its work force, management believes that it has sufficient human capital to operate its business successfully.
Competitive Pay and Benefits. Our compensation programs are designed to align the compensation of our employees with our performance and to provide the proper incentives to attract, retain and motivate employees to achieve superior results. The structure of our compensation programs balances incentive earnings for both short-term and long-term performance. Specifically:
| ● | We provide employee wages that are competitive and consistent with employee positions, skill levels, experience, knowledge and geographic location. |
| ● | Annual increases and incentive compensation are based on merit, which is communicated to employees at the time of hiring and documented through our talent management process as part of our annual review procedures and upon internal transfer and/or promotion. |
| ● | All full-time employees are eligible for health insurance, paid and unpaid leaves, a 401K retirement plan with employer matching contributions (maximum of 4% match), and life insurance coverage. We also offer a variety of voluntary benefits that allow employees to select the options that meet their needs, including flexible time-off, telemedicine, and paid parental leave. |
Health and Safety
The health and safety of our employees is our highest priority, and this is consistent with our operating philosophy. Accordingly, with the global spread of the ongoing novel coronavirus pandemic, we have implemented plans designed to address and mitigate the impact of the COVID-19 pandemic on the safety of our employees and our business, which include:
| ● | Adding work from home flexibility; |
| ● | Adjusting attendance policies to encourage those who are sick to stay home; |
| ● | Increasing cleaning protocols across all locations; |
| ● | Initiating regular communication regarding impacts of the COVID-19 pandemic, including health and safety protocols and procedures. |
Legal Proceedings
We are subject to claims and legal actions that arise in the ordinary course of business from time to time. However, we are not currently subject to any claims or actions that we believe would have a material adverse effect on our financial position or results of operations.
Properties
On March 1, 2020, we entered into a sublease with Purnovate, now our subsidiary and at the that time a related party, for the lease of three offices at 1180 Seminole Trail, Suite 495, Charlottesville, VA 22901. The lease has a term of two years, and the monthly rent is $1,400. The lease is terminable on thirty (30) days notice. On January 25, 2021, we acquired Purnovate. After the acquisition, the Company directly or through Purnovate operates a chemistry and analytics laboratory in its 4,175 square feet leased laboratory and office space. On January 6, 2020, Purnovate entered a lease for the Facility with a term of three (3) years. Included in the lease was the use of certain laboratory instrumentation and certain chemical assets. On January 19, 2021, Purnovate entered an amendment to this lease extending the lease until January 31, 2026.
On December 19, 2018, we entered into an office service agreement, which commenced on January 2, 2019, for two furnished workspaces (approximately 250 square feet) located at 1001 Research Park Blvd., Suite 100, Charlottesville, Virginia 22911. Pursuant to the agreement agreed to pay rent in the amount of $1,150 per month. Either party may terminate the sublease upon written notice to the other party specifying the date of termination as long as such date of termination is not earlier than the last day of the month following the month in which such notice is given.
On October 9, 2018, we entered into a license and membership agreement with Jelly Works X Zero-Ten, LLC for membership in a coworking space and use of an office located at 307A Kamani Street, Honolulu, HI 96813. We agreed to pay a monthly fee of $1,152 for membership and use of these facilities, committing to do so for a term of one year. The agreement is not a lease and does not create a tenancy relationship. At the end of this period, the agreement reverted to a month-to-month rental of a dedicated desk space, without office, for a monthly fee of $393 per month.
MANAGEMENT
Information About our Executive Officers and Directors
Our business and affairs are organized under the direction of our board of directors, which currently consists of six members.
In accordance with the terms of our certificate of incorporation, our board of directors is divided into three classes, as follows:
| ● | Class I, which will consist of William B. Stilley, III and Kevin Schuyler, whose term will expire at our annual meeting of stockholders to be held in 2022; |
| ● | Class II, which will consist of Tony Goodman and Robertson H. Gilliland, whose terms will expire at our annual meeting of stockholders to be held in 2023; and |
| ● | Class III, which will consist of J. Kermit Anderson and James W. Newman, Jr., whose terms will expire at our annual meeting of stockholders to be held in 2021. |
At each annual meeting of stockholders to be held after the initial classification, the successors to directors whose terms then expire will serve until the third annual meeting following their election and until their successors are duly elected and qualified. The authorized number of directors may be changed only by resolution of the board of directors. Any additional directorships resulting from an increase in the number of directors will be distributed between the three classes so that, as nearly as possible, each class will consist of one-third of the directors.
Set forth below are our directors and executive officers and their respective ages and positions as of March 22, 2021:
| Executive Officers and Directors | | Age | | Position(s) Held |
| William B. Stilley, III, MBA | | 53 | | Chief Executive Officer, President and Director |
| Joseph Truluck, MBA | | 42 | | Chief Operating Officer and Chief Financial Officer |
| Bankole A. Johnson, DSc, MD | | 61 | | Chief Medical Officer |
| Robertson H. Gilliland, MBA | | 40 | | Director |
| Tony Goodman | | 56 | | Director |
| J. Kermit Anderson | | 71 | | Director |
| James W. Newman, Jr. | | 78 | | Director |
| Kevin Schuyler, MBA, CFA | | 52 | | Director, Vice Chairman of the Board, Lead Independent Director |
There are no family relationships among any of our directors or executive officers. The executive officers and directors named above may act as authorized officers of the Company when so deemed by resolutions of the Company. Set forth below is a summary of the business experience of each of our directors and executive officers identified above and our key employee:
William B. Stilley, III, Chief Executive Officer, President and Director
William B. Stilley has served as our Chief Executive Officer since December 2010, our Secretary and Treasurer from April 2012 until October 2017 and a director since April 2011. In July 2018, Mr. Stilley was appointed to serve as a member of the board of directors of Avalon GloboCare Corp. (Nasdaq: AVCO), where he also serves as Chairman of the audit committee. Avalon GloboCare Corp. is a global intelligent biotech developer and healthcare service provider dedicated to promoting and empowering high impact, transformative cell-based technologies and their clinical applications, as well as healthcare facility management. Prior to joining the Company from August 2008 until December 2010, Mr. Stilley was the Vice President, Business Development & Strategic Projects at Clinical Data, Inc. (Nasdaq: CLDA). At Clinical Data, Inc., Mr. Stilley worked on licensing and M&A transactions and was involved in management of Phase 3 clinical trials, production of Viibryd ® for initial commercial launch of the product, and sourcing drug product and drug substance for the Phase 3 clinical trials of the company’s vasodilator drug for myocardial stress imaging. From February 2002, Mr. Stilley was the COO and CFO of Adenosine Therapeutics, LLC where he ran the internal operations of the company, including research and development, and all financing activity, until the sale of its principal assets were acquired by Clinical Data, Inc. in August 2008. Deals closed include, without limitation, financings, licenses or acquisition agreements with Johnson & Johnson, Novartis, Santen Pharmaceuticals, Epix Pharmaceuticals, CombinatoRx, ATEL Ventures, Medical Predictive Sciences Corporation, and Novartis Ventures. Mr. Stilley has advised both public and private companies on financing and M&A transactions, has been the interim CFO of a public company, the interim Chief Business Officer of Diffusion, and the COO and CFO of a number of private companies. Before entering the business community, Mr. Stilley served as Captain in the U.S. Marine Corps.
Mr. Stilley has an MBA with honors from the Darden School of Business and a B.S. in Commerce/Marketing from the McIntire School of Commerce at the University of Virginia. He has guest lectured at the Darden School of Business in two courses on the management of life science companies and, until recently, served on the board of directors of Virginia BIO, the statewide biotechnology organization.
We selected Mr. Stilley to serve on our board of directors because he brings to the board extensive knowledge of the biotechnology industry. Having served in senior corporate positions in several biomedical companies, he has a vast knowledge of the industry and brings to the board significant executive leadership and operational experience as well as knowledge and experience of financing and M&A transactions. His business experience provides him with a broad understanding of the operational, financial and strategic issues facing public companies and his extensive knowledge financing and M&A will serve our company well in the future.
Joseph Truluck, Chief Operating Officer, Chief Financial Officer, Treasurer and Secretary
Joseph Truluck has served as our Chief Operating Officer since April 2017, our Chief Financial Officer since June 2017, our Treasurer and Secretary since October 2017, and from May 2016 until his appointment as our Chief Operating Officer, as our VP Operations and Finance. Since January 2013, Mr. Truluck has served as the VP Operations and Finance at Adenosine Therapeutics, LLC after the company reacquired its major drug development program. As VP Operations and Finance, at Adenosine Therapeutics, Mr. Truluck has overseen the operations of the business, including seeing to completion a project to merge and analyze two partially completed Phase 3 trials to constitute a single trial. From April 2005 to July 2009, Mr. Truluck served as the Operations Manager of Adenosine Therapeutics’ until its purchase in August 2008 by Clinical Data. After the purchase of Adenosine Therapeutics’ operations by Clinical Data, Mr. Truluck went on to gain an MBA from Tulane University with a concentration in Finance. In addition to his MBA at Tulane, Mr. Truluck earned an MA in Philosophy at the University of Virginia, with a thesis in the area of modal semantics.
Bankole A. Johnson, D.Sc., M.D., Chief Medical Officer
Bankole Johnson has served as our Chief Medical Officer since March 24, 2019. Dr. Johnson also served as the Chairman of our Board from November 2010 until March 24, 2019. Dr. Johnson is a world-leading neuroscientist and a pioneer in the development of medications for the treatment of alcohol abuse and is the inventor of all patents covering AD04. In August 2013, he was appointed Chairman of the Department of Psychiatry at the University of Maryland School of Medicine and also leads the Brain Science Research Consortium Unit at the University of Maryland, a position he held until March, 2019 to devote greater focus to his new duties with us. Previously, from 2004 until August 2013, he served as Alumni Professor and Chairman of the Department of Psychiatry and Neurobehavioral Sciences at the University of Virginia.
Dr. Johnson graduated in Medicine from Glasgow University in 1982 and trained in Psychiatry at the Royal London and Maudsley and Bethlem Royal Hospitals. Additional to his medical degree, he trained in research at the Institute of Psychiatry (University of London) and conducted studies in neuropsychopharmacology for his doctoral thesis (degree from Glasgow University) on the Medical Research Council unit at Oxford University. In 2004, Dr. Johnson earned his Doctor of Science degree in Medicine from Glasgow University — the highest degree that can be granted in science by a British university. His primary area of research expertise is the psychopharmacology of medications for treating addictions.
Dr. Johnson is a licensed physician and board-certified psychiatrist throughout Europe and in the U.S. He is the Principal Investigator on National Institutes of Health (NIH)-funded research studies utilizing neuroimaging, neuropharmacology, and molecular genetics techniques. Dr. Johnson’s clinical expertise is in the fields of addiction, biological, and forensic psychiatry. Honors include service on numerous NIH review and other committees including special panels.
Dr. Johnson was the 2001 recipient of the Dan Anderson Research Award for his “distinguished contribution as a researcher who has advanced the scientific knowledge of addiction recovery.” He received the Distinguished Senior Scholar of Distinction Award in 2002 from the National Medical Association. Dr. Johnson also was an inductee of the Texas Hall of Fame in 2003 for contributions to science, mathematics, and technology, and in 2006 he received the American Psychiatric Association’s (APA’s) Distinguished Psychiatrist Lecturer Award. In 2007, he was named as a Fellow in the Royal College of Psychiatrists, and in 2008 he was elected to the status of Distinguished Fellow of the APA. In 2009, he received the APA’s Solomon Carter Fuller Award, honoring an individual who has pioneered in an area that has benefited significantly the quality of life for Black people. In 2010, he was named as a Fellow in the American College of Neuropsychopharmacology. Dr. Johnson is Field Editor -in-Chief of Frontiers in Psychiatry, serves on the Editorial Board of The American Journal of Psychiatry, and reviews for over 30 journals in pharmacology, neuroscience, and the addictions. He has over 200 publications. Dr. Johnson also has edited three books: Drug Addiction and Its Treatment: Nexus of Neuroscience and Behavior, Handbook of Clinical Alcoholism Treatment, and Addiction Medicine: Science and Practice, one of the foremost reference textbooks in the field.
Dr. Johnson has served as a consultant to Johnson & Johnson (Ortho-McNeil Janssen Scientific Affairs, LLC), Transcept Pharmaceuticals, Inc., D&A Pharma, Organon, Adial Corporation, Psychological Education Publishing Company (PEPCo LLC), and Eli Lilly and Company. He also has served on the Extramural Advisory Board for NIAAA (2004-present), the National Advisory Council for NIDA (2004-2007), the Medications Development Subcommittee of NIDA’s Advisory Council on Drug Abuse (2004-2007), and the Medications Development Scientific Advisory Board for NIDA (2005-2009). In addition, he has been the recipient of research grant support from both NIAAA and NIDA.
Robertson H. Gilliland, MBA, Director
Mr. Gilliland has served as a director since September 2014. Since May 2020, Mr. Gilliland has served as an independent consultant to family offices, with specific focus on investment strategy formulation and governance. From July 2013 until April 2020, he was Principal and Chief Financial Officer at Keller Enterprises, LLC, a family office that invests and manages private capital. In addition to his duties as CFO, as a principal, Mr. Gilliland sourced, vetted and managed a variety of private direct investments and spearheaded internal strategic initiatives. Prior to joining Keller Enterprises, Mr. Gilliland attended business school beginning in 2011 and was previously a Director at the Brunswick Group, where he specialized in strategic communications and investor relations around mergers and acquisitions, including being an advisor on the Pfizer-Wyeth, Celgene-Pharmion, and Mylan-Merck KGaA Generic transactions. During his tenure at Brunswick, Mr. Gilliland worked on over 35 multi-billion dollar M&A transactions. He has his MBA from the University of Michigan’s Ross School of Business, where he graduated with honors.
We selected Mr. Gilliland to serve on our board of directors because he brings extensive knowledge of the financial markets. Mr. Gilliland’s business background provides him with a broad understanding of the financial markets and the financing opportunities available to us.
Tony Goodman, Director
Tony Goodman has served as a director since July 2017. Mr. Goodman’s career spans over 23 years in Pharma and Biotech. Mr. Goodman is the Founder/Managing Director of Keswick Group, LLC, a Biotech Strategic Commercial and Business Development Advisory Firm. From October 2014 until February 2017, he served as the Chief Business Development Officer of Indivior PLC (INDV, FTSE 500) and a member of the executive team which brought Indivior public as a demerger from Reckitt Benckiser Pharmaceuticals, Inc. Mr. Goodman held many leadership positions at Reckitt Benckiser Pharmaceuticals from October 2009 until October 2014 that include: Global Director, Strategy and Commercial Development; Global Head, Category Development; and Director of US Commercial Managed Care. Mr. Goodman has also served as the Director of Strategic Marketing and Business Development at PRA International and Group Product Manager, Marketing and Director of the Managed Health Strategies Group at Purdue Pharmaceuticals L.P. Mr. Goodman graduated from Marshall University, with a degree in Business Administration and is currently a Full Board Executive with the National Association of Corporate Directors (“NACD”).
We selected Mr. Goodman to serve on our board of directors because he brings extensive knowledge of the addiction and pharmaceuticals industry and his significant strategic development experience. Mr. Goodman’s position at the NACD provides him with a broad understanding of the role of directors and corporate governance issues facing public companies.
J. Kermit Anderson, Director
J. Kermit Anderson has served as a director since February 2015. He has served as the VP and Chief Financial Officer at Cumberland Development Co. since 2007. Cumberland is a privately held company which evaluates and oversees investments in minerals exploration, life sciences, and real estate for a family office. Mr. Anderson has over forty years of experience in financial and development roles for a number of companies. He holds widely diversified experience in financial planning and reporting, accounting, forecasting, pricing, GAAP reporting and contract negotiations including benefits and compensation. His career is split almost equally between public and private companies including major sales and acquisitions. He has held various positions in energy businesses including Massey Energy, AMVEST and Cumberland Resources Corporation working on the sale of the companies for the last two roles. Mr. Anderson has worked extensively on startups for Massey and AMVEST including the move to a new business area with AMVEST. He received his BS -BA from West Virginia University in 1972.
We selected Mr. Anderson to serve on our board of directors because he brings extensive industry experience in corporate development and finance. His prior service with other public companies provides experience related to good corporate governance practices.
James W. Newman, Jr., Director
James W. Newman, Jr. has served as a director since September 2014. Since April 2013, he served as the Founder, Chairman, and President of Medical Predictive Science Corporation (“MPSC”), a medical device company that translates ICU research discoveries to the patient’s bedside and develops predictive technology that detects imminent, catastrophic illness. MPSC’s HeRO sold in over 20 countries and is a pioneering monitoring system for premature infants which detects early signs of distress commonly caused by infection and other potentially life -threatening illnesses. He has also served as part of the management team of Newman Company, a real estate company, since 1980, for which he still works and is the sole owner. In the mid -1990s he began making capital investments in several “start-up” companies, including Charlottesville-based Medical Automation Systems, a major provider of information management systems for point-of-care testing, which was acquired by Massachusetts-based Alere Inc. in 2011. His investments have covered a wide range of fields, encompassing everything from biotechnology, bio-informatics, education, and telecommunications, as well as mechanical inventions. He is particularly interested in investments in the medical field that improve healthcare, but do so at a reduced cost to consumers. Mr. Newman received a B.A. degree from Upsala College in 1968.
We selected Mr. Newman to serve on our board of directors because he brings a strong business background to our company and adds significant strategic, business and financial experience. Mr. Newman’s business and finance background provides him with a broad understanding of the issues faced by companies similar to us.
Kevin Schuyler, CFA – Director, Vice Chairman of the Board of Directors, Lead Independent Director
Kevin Schuyler has served as a director since April 2016 and is our Vice Chairman of the board of directors and Lead Independent Director. He currently serves as a senior managing director at CornerStone Partners, a full-service institutional CIO and investment office located in Charlottesville, VA, with approximately $10 billion under management. Prior to joining CornerStone Partners in 2006, he held various positions with McKinsey & Company, Louis Dreyfus Corporation and The Nature Conservancy. Mr. Schuyler serves on various boards and committees of Sentara Martha Jefferson Hospital, the US Endowment for Forestry and Communities, and Stone Barns Center. He is a member of the investment committee of the Margaret A. Cargill Philanthropies. Mr. Schuyler graduated with honors from Harvard College and received his MBA from The Darden Graduate School of Business at the University of Virginia. He is a member of the Chartered Financial Analyst Society of Washington, DC.
We selected Mr. Schuyler to serve on our board of directors because he brings extensive knowledge of the financial markets. Mr. Schuyler’s business background provides him with a broad understanding of the financial markets and the financing opportunities available to us.
Board Composition and Election of Directors
Our board of directors consists of six members: Messrs. Kermit Anderson, Robertson Gilliland, Tony Goodman, James Newman, Kevin Schuyler, and William Stilley. Our board of directors has undertaken a review of its composition and its committees and the independence of each director. Based upon information requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships, our board of directors has determined that each of Messrs. Kermit Anderson, Robertson Gilliland, Tony Goodman, James Newman, and Kevin Schuyler is “independent” under the applicable rules of the SEC and Nasdaq and that Mr. Stilley is not “independent” as defined under the such rules. In making such determination, our board of directors considered the relationship that each such non-employee director has with our company and all other facts and circumstances that our board of directors deemed relevant in determining his independence, including the beneficial ownership of our capital stock by each non-employee director. Mr. Stilley is not an independent director under these rules because he is our Chief Executive Officer and President.
Corporate Governance
Board Committees
Our board of directors has established an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee. From time to time, the Board of Directors may also establish ad hoc committees to address particular matters.
Audit Committee
The members of our Audit Committee are Messrs. Schuyler, Newman, and Goodman each of whom has been determined by our board of directors to be independent under applicable Nasdaq and SEC rules and regulations. Mr. Schuyler is the chair of the Audit Committee. Our Audit Committee’s responsibilities include, among others:
| ● | appointing, approving the compensation of, and assessing the independence of our registered public accounting firm; |
| ● | overseeing the work of our independent registered public accounting firm, including through the receipt and consideration of reports from that firm; |
| ● | reviewing and discussing with management and our independent registered public accounting firm our annual and quarterly financial statements and related disclosures; |
| ● | monitoring our internal control over financial reporting, disclosure controls and procedures; |
| ● | overseeing our internal audit function; |
| ● | discussing our risk management policies; |
| ● | establishing policies regarding hiring employees from our independent registered public accounting firm and procedures for the receipt and retention of accounting related complaints and concerns; |
| ● | meeting independently with our internal auditing staff, if any, our independent registered public accounting firm and management; |
| ● | reviewing and approving or ratifying any related person transactions; and |
| ● | preparing the Audit Committee report required by Securities and Exchange Commission, or SEC, rules. |
All audit and non-audit services, other than de minimis non-audit services, to be provided to us by our independent registered public accounting firm must be approved in advance by our Audit Committee.
Our board of directors has determined that Mr. Schuyler is an “audit committee financial expert” as defined in applicable SEC rules.
Compensation Committee
The members of our Compensation Committee are Messrs. Anderson and Newman, each of whom has been determined by our board of directors to be independent under current Nasdaq rules and regulations. Mr. Anderson is the chair of the Compensation Committee. Our Compensation Committee’s responsibilities include, among others:
| ● | reviewing and approving annually the corporate goals and objectives applicable to the compensation of the Chief Executive Officer, evaluating at least annually the Chief Executive Officer’s performance in light of those goals and objectives, and determining and approving the Chief Executive Officer’s compensation level based on this evaluation; |
| ● | reviewing and approving the compensation of all other executive officers; |
| ● | reviewing and approving and, when appropriate, recommending to the board of directors for approval, incentive compensation plans and equity-based plans, and where appropriate or required, recommending for approval by the stockholders of the Company, the adoption, amendment or termination of such plans; and administering such plans; |
| ● | reviewing and approving the executive compensation information included in our annual report on Form 10-K and proxy statement; |
| ● | reviewing and approving or providing recommendations with respect to any employment agreements or severance arrangements or plans; and |
| ● | reviewing director compensation and recommending any changes to the board of directors. |
Nominating and Corporate Governance Committee
The members of our Nominating and Corporate Governance Committee are Messrs. Gilliland, and Goodman, each of whom has been determined by our board of directors to be independent under current Nasdaq rules. Mr. Gilliland is the chair of the Nominating and Corporate Governance Committee. Our Nominating and Corporate Governance Committee’s responsibilities include, among others:
| ● | identifying and recommending candidates to fill vacancies on the board of directors and for election by the stockholders; |
| ● | recommending committee and chairperson assignments for directors to the board of directors; |
| ● | developing, subject to the board of directors’ approval, a process for an annual evaluation of the board of directors and its committees and to oversee the conduct of this annual evaluation; |
| ● | overseeing the Company’s corporate governance practices, including reviewing and recommending to the board of directors for approval any changes to the documents and policies in the Company’s corporate governance framework, including its certificate of incorporation and bylaws; and |
| ● | monitoring compliance with the Company’s Code of Business Conduct and Ethics, investigating alleged breaches or violations thereof and enforcing its provisions. |
Special Committee
During 2020,the Board of Directors also formed a Special Committee comprised of Tony Goodman, Robin Gilliland and until his transition as a director, Jack Reich, in order to negotiate and consummate the Acquisition of Purnovate, which Special Committee terminated after the consummation of the Acquisition. Mr. Goodman and Mr. Gilliland each received cash compensation of $7,000 for serving on the Special Committee.
Board of Directors Leadership Structure
We currently have a separate lead independent director. Our lead independent director is Kevin Schuyler. In that role, he presides over the executive sessions of the board of directors, during which our independent directors meet without management, and he serves as the principal liaison between management and the independent directors of the board of directors. We do not have a formal policy regarding having a separate lead independent director. Our board of directors has determined its leadership structure is appropriate and effective for us, given our stage of development.
Risk Oversight
Our board of directors monitors our exposure to a variety of risks through our Audit Committee. Our Audit Committee charter gives the Audit Committee responsibilities and duties that include discussing with management, the internal audit department and the independent auditors our major financial risk exposures and the steps management has taken to monitor and control such exposures, including our risk assessment and risk management policies.
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics that applies to all of our employees, officers, and directors, including those officers responsible for financial reporting. These standards are designed to deter wrongdoing and to promote honest and ethical conduct. The code of business conduct and ethics and the written charter for the audit committee, compensation committee and nominating and corporate governance committee are available on our website. The information that appears on our website is not part of, and is not incorporated into, this registration statement.
None of our directors or executive officers, nor any associate of such individual, is involved in a legal proceeding adverse to us.
If we make any substantive amendments to the code of business conduct and ethics or grant any waiver from a provision of the code to any executive officer or director, we will promptly disclose the nature of the amendment or waiver on our website. We will promptly disclose on our website (i) the nature of any amendment to the policy that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and (ii) the nature of any waiver, including an implicit waiver, from a provision of the policy that is granted to one of these specified individuals, the name of such person who is granted the waiver and the date of the waiver.
EXECUTIVE COMPENSATION
Summary Compensation Table
The following table sets forth the information as to compensation paid to or earned by our executive officers during the years ended December 31, 2020 and 2019 whose total compensation did exceed $100,000. The persons listed in the following table are referred to herein as the “named executive officers.”
| Name and Principal Position | | Fiscal
Year | | Salary | | | Bonuses | | | Option
Award(s) | | | All Other
Compensation | | | Total | |
| William B. Stilley | | 2020 | | $ | 400,000 | | | $ | 150,000 | (1) | | $ | 519,800 | (2) | | $ | 64,103 | (3) | | $ | 1,133,903 | |
| Chief Executive Officer and Member of the board of directors | | 2019 | | $ | 381,695 | | | $ | 620,000 | (4) | | $ | 1,311,509 | (5) | | $ | 53,183 | (6) | | $ | 2,366,387 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Joseph A. M. Truluck | | 2020 | | $ | 167,493 | | | $ | 50,000 | (7) | | $ | 226,000 | (8) | | $ | 7,540 | (9) | | $ | 451,033 | |
| Chief Operating Officer and Chief Financial Officer | | 2019 | | $ | 144,931 | | | $ | 110,000 | (10) | | $ | 472,143 | (11) | | $ | – | | | $ | 727,074 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Bankole A. Johnson | | 2020 | | $ | 375,000 | | | $ | – | | | $ | – | | | $ | – | | | $ | 375,000 | |
| Chief Medical Officer(12) | | 2019 | | $ | 271,673 | | | $ | 250,000 | (13) | | $ | 581,575 | (14) | | $ | 6,332 | (15) | | $ | 1,109,580 | |
| (1) | Bonuses for Mr. Stilley were comprised of cash performance bonus payment of $120,000 earned in 2020 and paid in 2021 with an additional $30,000 extraordinary cash bonus paid in 2021. |
| (2) | Includes the fair value of 460,000 options to purchase shares of common stock at an exercise price of $1.44 per share issued on March 2, 2020 at a fair value of $1.13 per option. Options vest over a three year period from grant date. Fair value computed in accordance with FASB ASC Topic 718. |
| (3) | All other compensation for Mr. Stilley is comprised of (i) a contribution by our company to an HSA ($8,004); (ii) the payment by our company of insurance premiums including life, dental, vision ($28,086); (iii) $8,013 in matched 401(k) contributions; and (iv) cash fee for services as a Director ($20,000). |
| (4) | Bonuses for Stilley are comprised of (i) cash extraordinary performance bonus payment of $500,000 in 2019; and (ii) $120,000 in bonus payments earned in 2019, paid in 2020 with $42,000 in cash and $78,000 in restricted stock grants. |
| (5) | Represents the fair value of 500,000 options to purchase shares of common stock at an exercise price of $3.39 per share issued on March 10, 2019 at a fair value of approximately $2.62 per option. Options vest over a three year period from grant date. Fair value computed in accordance with FASB ASC Topic 718. |
| (6) | All other compensation for Mr. Stilley is comprised of (i) a contribution by our company to an HSA ($8,004); (ii) the payment by our company of insurance premiums including life, dental, vision ($25,179); and (iii) cash fee for services as a Director ($20,000). |
| (7) | Consisting of a cash performance bonus payment of $34,000 fully earned in 2020 and an additional $16,000 extraordinary performance cash bonus paid in 2021. |
| (8) | Represents the fair value of 200,000 options to purchase shares of common stock at an exercise price of $1.44 per share issued on March 3, 2020 at a fair value of approximately $1.13 per option. Options vest over a three year period from grant date. Fair value computed in accordance with FASB ASC Topic 718. |
| (9) | Comprised of $7,540 in matched 401(k) contributions. |
| (10) | Consisting of a cash performance bonus payment of $50,000 in 2019 and $60,000 in bonus payments earned in 2019, paid in 2020 with $21,000 in cash and $39,000 in restricted stock grants. |
| (11) | Represents the fair value of 180,000 options to purchase shares of common stock at an exercise price of $3.39 per share issued on March 10, 2019 at a fair value of approximately $2.62 per option. Options vest over a three year period from grant date. Fair value computed in accordance with FASB ASC Topic 718. Also includes the fair value of 27,084 shares common stock issued on March 3, 2020 at a market price of $1.44. |
| (12) | Dr. Johnson serves in the capacity of Chief Medical Officer as an independent contractor, and those amounts disclosed as salary represent consulting fees paid him for his services. |
| (13) | Consisting of a $250,000 signing bonus on execution of Dr. Johnson’s consulting agreement. |
| (14) | Represents the fair value of 250,000 options to purchase shares of common stock at an exercise price of $3.01 per share issued on March 10, 2019 at a fair value of approximately $2.33 per option. Options vest over a three year period from grant date. Fair value computed in accordance with FASB ASC Topic 718. |
| (15) | Consists of $6,332 in compensation as Chairman of the Board of Directors prior to assuming the position of CMO. (This amount was disclosed in previous years under Director Compensation.) Compensation paid to Dr. Johnson as a result of a vendor agreement with PEPCO, a company owned by Dr. Johnson, is not included in this table, including unrestricted stock grant worth $4,812 and cash payments of $24,251 made to PEPCO for services provided in the fourth quarter of 2019, and a cash payment of $219,823 made to PEPCO in advance of services provided on December 12, 2019. |
Outstanding Equity Awards at Fiscal Year-End (December 31, 2020)
The following table provides information about the number of outstanding equity awards held by each of our named executive officers as of December 31, 2020:
| Option Awards | | Stock Awards | |
| Name | | Number of
Securities
Underlying
Unexercised
Options
(Exercisable) | | | Number of
Securities
Underlying
Unexercised
Options
(Unexercisable) | | | Option
Exercise
Price | | | Option
Expiration
Date | | Equity
Incentive
Plan Awards:
Number of
Unearned
Shares That
Have Not
Vested | | | Equity
Incentive
Plan
Awards:
Market or
Payout Value
of Unearned
Shares That
Have Not
Vested | |
| William B. Stilley | | | 57,471 | | | | – | (1) | | $ | 5.70 | | | 6/30/2027 | | | | | | | | |
| Chief Executive Officer and Member of the Board of Directors | | | 305,556 | | | | 194,444 | (2) | | $ | 3.39 | | | 3/9/2029 | | | | | | | | |
| | | | 127,778 | | | | 332,222 | (3) | | $ | 1.44 | | | 3/3/2030 | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Joseph Truluck | | | 30,132 | | | | – | (1) | | $ | 5.70 | | | 6/30/2027 | | | | | | | | |
| Chief Operating Officer and Chief Financial Officer | | | 110,000 | | | | 70,000 | (2) | | $ | 3.39 | | | 3/9/2029 | | | | | | | | |
| | | | 55,556 | | | | 144,444 | (3) | | $ | 1.44 | | | 3/3/2030 | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Bankole A. Johnson | | | 5,580 | | | | – | (1) | | $ | 5.70 | | | 6/30/2027 | | | | | | | | |
| Chief Medical Officer | | | 152,778 | | | | 97,222 | (4) | | $ | 3.01 | | | 3/24/2029 | | | | | | | | |
| (1) | One thirty-sixth (1/36) of these options vested on the date of grant, June 30, 2017, with an additional one thirty-sixth vesting on the first day of each subsequent month. |
| (2) | One thirty-sixth (1/36) of these options vested on the date of grant, March 9, 2019, with an additional one thirty-sixth vesting on the first day of each subsequent month. |
| (3) | One thirty-sixth (1/36) of these options vested on the date of grant, March 3, 2020, with an additional one thirty-sixth vesting on the first day of each subsequent month. |
| (4) | One thirty-sixth (1/36) of these options vested on the date of grant, March 25, 2019, with an additional one thirty-sixth vesting on the first day of each subsequent month. |
Does not include the grant on February 8, 2021 of an option to each of William B. Stilley and Joseph Truluck of an option to purchase 250,000 and 125,000 shares of our common stock, respectively. The shares of common stock underlying the option awards each vest pro rata on a monthly basis over a thirty-six month period.
Employment Agreements and Consulting Agreement
Employment Agreements
We are currently a party to employment agreements with each of Messrs. Stilley and Truluck.
Effective upon the closing of our initial public offering, we entered into a five -year employment agreement with Mr. Stilley to continue to serve as our Chief Executive Officer, which agreement was amended on February 12, 2021 to extend the term of the agreement to March 31, 2026 (the “Stilley EA”). Under the Stilley EA, as amended on March 10, 2019 to increase his salary to $400,000 and further amended on February 12, 2021 and March 17, 2021, Mr. Stilley will receive an annual salary of $410,000 and has a target bonus opportunity equal to 40% of his salary. Mr. Stilley’s annual salary will be subject to increase at the discretion of our board of directors. Our board of directors may, in its discretion, pay a portion of Mr. Stilley’s annual bonus in the form of equity or equity -based compensation, provided that commencing with the year following the year in which a Change of Control (as defined in the Stilley EA) occurs, Mr. Stilley’s annual bonus will be paid in cash. Mr. Stilley will also subject to certain restrictive covenants, including a non -competition (applicable during employment and for 24 months thereafter), customer non -solicitation and employee and independent contractor non -solicitation (each applicable during employment and for 12 months thereafter), as well as confidentiality (applicable during employment and 7 years thereafter) and non -disparagement restrictions (applicable during employment and at all times thereafter).
Effective upon the closing of the initial public offering, we entered into a three -year employment agreement with Joseph Truluck to serve as our Chief Operating Officer and Chief Financial Officer (the “Truluck EA”), which agreement was amended on February 12, 2021 to extend the term of the agreement to March 31, 2026. Under the Truluck EA, Mr. Truluck devotes no less than 50% of his business time to the affairs of our company, which was increased to 75% on February 12, 2021. Pursuant to the terms of the Truluck EA, as amended on March 10, 2019 to increase his salary to $150,000 per annum and further amended on March 3, 2020 to increase his salary to ($170,000 per annum) and further amended on February 8, 2021, he receives an annual salary of $260,000 and has a target bonus opportunity equal 25% of his salary. Mr. Truluck’s annual salary is subject to increase at the discretion of our board of directors. Our board of directors may, in its discretion, pay a portion of Mr. Truluck’s annual bonus in the form of equity or equity -based compensation. Mr. Truluck is also subject to certain restrictive covenants, including a non -competition (applicable during employment and for 24 months thereafter), customer non -solicitation and employee and independent contractor non -solicitation (each applicable during employment and for 12 months thereafter), as well as confidentiality (applicable during employment and 7 years thereafter) and non -disparagement restrictions (applicable during employment and at all times thereafter).
In the event that Mr. Stilley’s or Mr. Truluck’s (each an “Executive”) employment is terminated by us other than for Cause, or upon his resignation for Good Reason (as such terms are defined in the Employment Agreement), the Executive will be entitled to any unpaid bonus earned in the year prior to the termination, a pro -rata portion of the bonus earned during the year of termination, continuation of base salary for 12 months for Mr. Stilley and 6 months in the case of Mr. Truluck, plus 12 months of COBRA premium reimbursement. If Mr. Stilley’s termination occurs within 60 days before or within 24 months following a Change of Control, then Mr. Stilley will be entitled to receive.
The same severance benefits as provided above except he will receive (a) a payment equal to two times the sum of his base salary and the higher of his target annual bonus opportunity and the bonus payment he received for the year immediately preceding the year in which the termination occurred instead of 12 months of base salary continuation and (b) 24 times the monthly COBRA premium for himself and his eligible dependents instead of 12 months of COBRA reimbursements (the payments in clauses (a) and (b) are paid in a lump sum in some cases and partly in a lump sum and partly in installments over 12 months in other cases). In addition, if Mr. Stilley’s employment is terminated by us without Cause or by the Executive for Good Reason, in either case, upon or within 24 months following a Change of Control, then the Executive will be entitled to full vesting of all equity awards received by the Executive from us (with any equity awards that are subject to the satisfaction of performance goals deemed earned at not less than target performance).
In the event that the Executive’s employment is terminated due to his death or Disability, the Executive (or his estate) will be entitled to any unpaid bonus earned in the year prior to the termination, a pro -rata portion of the bonus earned during the year of termination, 12 months of COBRA premium reimbursement and accelerated vesting of (a) all equity awards received in payment of base salary or an annual bonus and (b) with respect to any other equity award, the greater of the portion of the unvested equity award that would have become vested within 12 months after the termination date had no termination occurred and the portion of the unvested equity award that is subject to accelerated vesting (if any) upon such termination under the applicable equity plan or award agreement (with performance goals deemed earned at not less than target performance, and with any equity award that is in the form of a stock option or stock appreciation right to remain outstanding and exercisable for 12 months following the termination date or, if longer, such period as provided under the applicable equity plan or award agreement (but in no event beyond the expiration date of the applicable option or stock appreciation right).
All severance payments to the Executives will be subject to the execution and non -revocation of a release of claims by the Executive or his estate, as applicable.
For purpose of each of the Stilley EA and Truluck EA, “Good Reason” is defined as the occurrence of any of the following events without the respective Executive’s consent: (i) a material reduction in the Executive’s duties, responsibilities or authority; (ii) a reduction of the Executive’s base salary; (iii) failure or refusal of a successor to us to either materially assume our obligations under the employment agreement or enter into a new employment agreement with the Executive on terms that are materially similar to those provided under this Agreement, in any case, in the event of a Change of Control; (iv) relocation of the Executive’s primary work location that results in an increase in the Executive’s one -way driving distance by more than twenty -five (25) miles from the Executive’s then -current principal residence; or (v) a material breach of the employment agreement by us.
For purposes of the Stilley EA and Truluck EA, “Cause” is defined as that the Executive shall have engaged in any of the following acts or that any of the following events shall have occurred, all as determined by the board of directors in its sole and absolute discretion: (i) conviction for, or entering of a plea of guilty or nolo contendere (or its equivalent under any applicable legal system) with respect to (A) a felony or (B) any crime involving moral turpitude; (ii) commission of fraud, misrepresentation, embezzlement or theft against any person; (iii) engaging in any intentional activity that injures or would reasonably be expected to injure (monetarily or otherwise), in any material respect, the reputation, the business or a business relationship of the Company or any of its affiliates; (iv) gross negligence or willful misconduct in the performance of the Executive’s duties to us or its affiliates under this Agreement, or willful refusal or failure to carry out the lawful instructions of the board of directors that are consistent with the Executive’s title and position; (v) violation of any fiduciary duty owed to us or any of its affiliates; or (vi) breach of any restrictive covenant (as defined) or material breach or violation of any other provision of the employment agreement, of a written policy or code of conduct of our company or any of our affiliates (as in effect from time to time) or any other agreement between the Executive and we or any of our affiliates. Except when such acts constituting Cause which, by their nature, cannot reasonably be expected to be cured, the Executive will have twenty (20) days following the delivery of written notice by the Company of its intention to terminate the Executive’s employment for Cause within which to cure any acts constituting Cause. Following such twenty (20) day cure period, and if the reason stated in the notice is not cured, the Executive shall be given five (5) business days prior written notice to appear (with or without counsel) before the full Board for the opportunity to present information regarding his views on the alleged Cause event. After we provide the original notice of our intent to terminate Executive’s employment for Cause, we may suspend the Executive, with pay, from all his duties and responsibilities and prevent him from accessing our or our affiliates premises or contacting any of our personal or any of our affiliates until a final determination on the hearing is made. The Executive will not be terminated for Cause until a majority of the independent directors approve such termination following the hearing.
For the purposes of each of the Stilley EA and Truluck EA, “Change in Control” is defined as: (i) the accumulation over a twelve (12) month period, whether directly or indirectly, by any individual, entity or group of our securities representing over fifty (50%) percent of the total voting power of all our then outstanding voting securities; (ii) a merger or consolidation of us in which our voting securities immediately prior to the merger or consolidation do not represent, or are not converted into securities that represent, a majority of the voting power of all voting securities of the surviving entity immediately after the merger or consolidation; (iii) a sale of substantially all of our assets; or (iv) during any period of twelve (12) consecutive months, our current directors, together with any new director whose election by the board of directors or nomination for election by the Company’s stockholders was approved by a vote of at least a majority of the directors then still in office, cease for any reason to constitute at least a majority of the board of directors.
Consulting Agreement
On March 24, 2019, we entered into a three-year consulting agreement with Bankole Johnson. Dr. Johnson’s consulting agreement with us (the “Consulting Agreement”) provides that Dr. Johnson will serve as our Chief Medical Officer and devote 75% of his working time to our business and affairs and will receive: (i) an annual fee of $375,000 a year; (ii) a signing bonus of $250,000 (which he received); and (iii) an option to purchase 250,000 shares of our common stock. The shares of common stock underlying the option award vests pro rata on a monthly basis over a thirty-six month period. The options are exercisable for a period of ten years from the date of grant and have an exercise price of $3.01 per share.
The Consulting Agreement may be terminated by us upon Dr. Johnson’s death, upon thirty days’ notice for a material breach of the Consulting Agreement by Dr. Johnson that can be cured, after notice of breach and failure to cure; upon notice for a breach of the Consulting Agreement by Dr. Johnson that cannot be cured; upon thirty days’ notice for any other cause; or upon thirty days’ notice (but not before 12 months from the effective date of the Consulting Agreement) at any time without cause; provided that if terminated by us without cause then Dr. Johnson will be entitled to receive his monthly payments for an additional six (6) months and his options will continue to vest for an additional six (6) months from the effective date of the notice of termination, subject to the terms of the 2017 Incentive Plan and the option agreement that we entered into with Dr. Johnson. In the event that Dr. Johnson’s termination is without cause and occurs within three months before or after a Significant Investment Event (as defined below), Dr. Johnson will be entitled to a buy -out payment in an amount equal to $31,250 times the number of months remaining on the initial term of the consulting agreement as of the effective date of the termination, minus the payment of the six (6) months of monthly payments provided for above (in addition to the immediate vesting at the time of termination of all remaining shares of our common stock or options to purchase shares of our common stock that would have otherwise.
Indemnification Agreements
We entered into agreements with each Executive and each director under which we will be required to indemnify them against expenses, judgments, damages, liabilities, losses, penalties, excise taxes, fines and amounts paid in settlement and other amounts actually and reasonably incurred in connection with an actual or threatened proceeding if any of them may be made a party because the Executive or director is or was one of our Executives. We will be obligated to pay these amounts only if the executive or director acted in good faith and in a manner that he or she reasonably believed to be in or not opposed to our best interests. With respect to any criminal proceeding, we will be obligated to pay these amounts only if the Executive or director had no reasonable cause to believe his/her conduct was unlawful. The indemnification agreements also set forth procedures that will apply in the event of a claim for indemnification.
Director Compensation
Director Compensation Table
The following table sets forth information regarding the compensation earned for service on our board of directors by our non-employee directors during the year ended December 31, 2020. Mr. Stilley also served on our board of directors, and received compensation as a result. The compensation for Mr. Stilley as an executive officer and Director is set forth above under “—Summary Compensation Table.”
| (a) Name | | (b)
Fees Earned or Paid
in Cash
($) | | | (c)
Stock
Awards
($) | | | (d)
Option
Awards(1)
($) | | | (e)
Non-Equity
Incentive
Plan
Compensation
($) | | | (f)
Change in
Pension Value
and
Nonqualified
Deferred
Compensation
Earnings
($) | | | (g)
All Other
Compensation
($) | | | (h)
Total
($) | |
| J. Kermit Anderson | | $ | 30,000 | | | | – | | | $ | 67,187 | | | $ | – | | | | – | | | | – | | | $ | 97,187 | |
| Robertson H. Gilliland, MBA | | $ | 34,000 | | | | – | | | $ | 67,187 | | | $ | – | | | | – | | | | – | | | $ | 101,187 | |
| Tony Goodman | | $ | 36,000 | | | | – | | | $ | 67,187 | | | $ | – | | | | – | | | | – | | | $ | 103,187 | |
| Jack W. Reich(2) | | $ | 17,006 | | | | – | | | $ | 104,531 | | | $ | – | | | | – | | | | – | | | $ | 121,531 | |
| James W. Newman, Jr. | | $ | 31,000 | | | | – | | | $ | 67,187 | | | $ | – | | | | – | | | | – | | | $ | 98,187 | |
| Kevin Schuyler, MBA, CFA | | $ | 35,000 | | | | – | | | $ | 67,187 | | | $ | – | | | | – | | | | – | | | $ | 102,187 | |
| (1) | As of December 31, 2020, the following are the total outstanding number of option awards held by each of our non-employee directors, all awards having been made prior to January 1, 2021: |
| Name | | Option
Award
(#) | |
| J. Kermit Anderson | | | 65,580 | |
| Robertson H. Gilliland, MBA | | | 65,580 | |
| Tony Goodman | | | 71,160 | |
| Jack W. Reich(2) | | | 90,000 | |
| James W. Newman, Jr. | | | 65,580 | |
| Kevin Schuyler, MBA, CFA | | | 65,580 | |
| (2) | Mr. Reich joined the Board of Directors on May 19, 2020 and left the Board to become VP, Regulatory on December 14, 2020. |
Directors receive cash compensation for their service as directors, including service as members of each committee on which they serve.
Does not include options to purchase 40,000 shares of our common stock issued to each non-executive director on February 10, 2021. The option vest pro rata on a monthly basis over 36 months.
On June 30, 2017, the board of directors approved a plan for the annual cash compensation of directors, which plan was amended on April 1, 2018 with respect to the Chairman’s compensation, which plan remained in effect in 2020:
| | | Board | | | Audit
Committee | | | Compensation
Committee | | | Nominating &
Governance
Committee | |
| Chair | | $ | 49,000 | | | $ | 15,000 | | | $ | 10,000 | | | $ | 7,000 | |
| Member | | $ | 20,000 | | | $ | 6,000 | | | $ | 5,000 | | | $ | 3,000 | |
On February 12, 2021, the Compensation Committee approved a plan for the annual cash compensation of directors for 2021:
| | | Board | | | Audit
Committee | | | Compensation
Committee | | | Nominating &
Governance
Committee | |
| Chair | | $ | 30,000 | | | $ | 16,000 | | | $ | 11,000 | | | $ | 8,000 | |
| Member | | $ | 24,000 | | | $ | 8,000 | | | $ | 6,000 | | | $ | 4,000 | |
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Related-Party Transactions
The following is a summary of transactions since January 1, 2019 to which we have been a party in which the amount involved exceeded the lesser of $120,000 or one percent of the average of our total assets at the end of the last two recent fiscal years and in which any of our executive officers, directors, director nominees or beneficial holders of more than five percent of our capital stock had or will have a direct or indirect material interest, other than compensation arrangements which are described under the section of this registration statement entitled “Management—Non-Employee Director Compensation” and “Executive Compensation.”
PEPCO MSA
On July 5, 2019, we entered into a Master Services Agreement (the “MSA”) and attached statement of work (the “SOW”) with Psychological Education Publishing Company (“PEPCO”) to administer a behavioral therapy program during our currently ongoing Phase 3 clinical trial using AD04, for the treatment of alcohol use disorder. Specifically, PEPCO is engaged in the business of training and certifying clinical investigators in the administration of Brief Behavioral Compliance Enhancement Treatment (“BBCET”). PEPCO is owned by Dr. Bankole Johnson, our Chief Medical Officer, and currently our largest stockholder. We may terminate the MSA at any time upon ten (10) days prior written notice to PEPCO. Unless otherwise indicated in our notice of termination, Work (as defined in the MSA) under any statement of work in progress at the time of the delivery of notice of termination shall continue as if the applicable statement of work had not been terminated, and the terms hereof shall continue to apply to such work. We may also terminate the MSA for cause due to PEPCO’s failure to perform its obligations thereunder upon three (3) days prior written notice to PEPCO; provided, however, the Company may terminate the MSA immediately in the event of PEPCO’s violation, or threatened violation, of certain provisions contained therein.
The statement of work under the MSA will terminate upon the completion the final study report for the Trial and delivery of the final report by PEPCO on the supervision and monitoring of the BBCET, including, without limitation, data reports. Notwithstanding the forgoing, the statement of work may be terminated by us upon written notice to PEPCO.
Prior to amendment (see below), it was anticipated that the compensation to be paid to PEPCO for services under the MSA would be approximately $300,000, of which subject to approval of the Nasdaq Capital Market shares of our common stock having a value equal to twenty percent (20%) of the fees due thereunder (the “Company Shares”) would be issued to Dr. Johnson as a consultant under the 2017 Equity Incentive Plan. On October 2, 2019, the Company issued 3,187 shares of common stock to Dr. Johnson at a market price of $1.51 per share and total value of $4,812 under the terms of the MSA.
On December 12, 2019, we entered into an Amendment (the “Amendment”) to the SOW. We had paid PEPCO $39,064 under the SOW for services rendered to date, leaving as estimated balance of $274,779 estimated to be paid under the SOW. The Amendment provided us with a 20% discount on the remaining services to be provided under the SOW and fixed the price of any remaining services under the SOW to be a total of $219,823 for all services required for the use of Brief Behavioral Compliance Enhancement Treatment (BBCET) in support of our ONWARD Phase 3 clinical trial provided that payment be made no later than December 13, 2019, which payment was made.
In addition, Dr. Johnson executed a guaranty, dated December 12, 2019, of PEPCO’s performance under the MSA and SOW (the “Guaranty”), together with a pledge and security agreement, dated December 12, 2019 (the “Pledge and Security Agreement”), to secure the Guaranty with 600,000 shares of our common stock beneficially owned by him and a lock-up agreement, dated December 12, 2019 (the “Lock-Up”), pursuant to which he agreed not to transfer or dispose of, directly or indirectly, any shares of our common stock, as currently owned by him, until after January 1, 2021.
On August 19, 2020, we and Dr. Bankole Johnson entered into a Lock-Up Agreement Extension and Right of First Refusal (the “Lock-Up Extension”), which amended the Lock-Up Agreement that had been entered into dated December 12, 2019 (the “Lock-Up”). The Lock-Up Extension extended the term of Dr. Johnson’s Lock-Up from January 1, 2021 until April 1, 2021. In connection with the Lock-Up Extension, Dr. Johnson was released from his Lock-Up restrictions with respect to 350,000 shares of our common stock, in order to enable Dr. Johnson to fund his new clinic focused on brain wellness and addiction treatments, Privée Clinics, LLC. Additionally, under the Lock-Up Extension, we were granted a right of first refusal for future financings in Privée Clinics, LLC.
Medical Translation Services Agreement
On January 29, 2018, we entered a Medical Translation Services Agreement with a firm controlled by Dr. Johnson. Under this agreement, the firm is responsible for translating our allowed patent for validation in 22 countries in Europe that require translation into the native language. In return for these services, we agreed to pay the firm $67,304 in installments through April 2018 and issue shares of our common stock upon consummation of our initial public offering or any other the sale by us of our equity securities resulting in gross proceeds of $2,000,000 or more with the stock to be issued valued at $201,911 based on the price per share of the common stock in such offering. During 2018, we paid the firm controlled by Dr. Johnson a total of $68,540 and upon consummation of our initial public offering, we issued such firm 40,463 shares of our common stock in full payment of all amounts owed under this agreement.
Grant Incentive Plan
On April 1, 2018, the board of directors approved and then revised, respectively, a Grant Incentive Plan to provide incentive for Bankole A. Johnson (the “Plan Participant”), to secure grant funding for us. Under the Grant Incentive Plan, we will make a cash payment to the Plan Participant each year based on the grant funding received by us in the preceding year in an amount equal to 10% of the first $1 million of grant funding received and 5% of grant funding received in the preceding year above $1 million. Amounts to be paid to the Plan Participants will be paid to each as follows: 50% in cash and 50% in stock. As of December 31, 2020, no grant funding that would result in a payment to the Plan Participant had been obtained.
Purnovate
On January 25, 2021, we closed the Acquisition contemplated by that Equity Purchase Agreement pursuant to which we purchased all of the outstanding membership interests of Purnovate from the members of Purnovate, such that after the Acquisition, Purnovate became our wholly owned subsidiary. Purnovate is a drug development company with a platform focused on developing drug candidates for non-opioid pain reduction and other diseases and disorders potentially targeted with adenosine analogs that are selective, potent, stable, and soluble.
William B. Stilley, our President and Chief Executive Officer and a member of its board of directors, and James W. Newman, a member of our board of directors, were Members or Purnovate and received $100,554 and $1,865 of the cash consideration and 201,109 and 3,731 shares of our common stock from the Acquisition. As previously stated, Mr. Stilley is subject to a two (2) year lock up with respect to the sale and transfer of the Stock Consideration that he received so long as his employment has not been terminated by us without cause prior to the end of such two (2) year period. Mr. Stilley owns approximately 28.7% of the membership interest of Purnovate and Mr. Newman controls two entities that, together, own less than 1% of the membership interests of Purnovate.
Purnovate Sublease
On March 1, 2020, the Company entered into a sublease with Purnovate, LLC, a private company in which our CEO has a 35% financial interest, for the lease of three offices at 1180 Seminole Trail, Suite 495, Charlottesville, VA 22901. The lease has a term of two years, and the monthly rent is $1,400.
Effective December 14, 2020, we appointed Dr. Jack Reich, who was then a director serving on our Board of Directors, as Head of Regulatory pursuant to an Offer Letter, dated December 14, 2020 (the “Reich Offer Letter”). Pursuant to the Reich Offer Letter, Dr. Reich served as Head of Regulatory, report directly to our Chief Executive Officer and was to receive a salary of $225,000 per year. Dr. Reich was also eligible for an annual bonus with a target of 30% of his salary, subject to the discretion of the Company’s board of directors.
PRINCIPAL STOCKHOLDERS
The following table sets forth certain information, as of March 22, 2021, with respect to the beneficial ownership of our common stock by each of the following:
| ● | each person who is known by us to be the beneficial owner of more than 5% of our outstanding common stock; |
| ● | each of our named executive officers; and |
| ● | all of our directors and executive officers as a group. |
As of March 22, 2021, we had 17,269,877 shares of common stock outstanding.
We have determined beneficial ownership in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of common stock issuable pursuant to the exercise of profits interest units, warrants or other rights that are either immediately exercisable or exercisable on or before May 21, 2021, which is approximately 60 days after the date of this prospectus. These shares are deemed to be outstanding and beneficially owned by the person holding those options or warrants for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them, subject to applicable community property laws.
Except as otherwise noted below, the address for each of the individuals and entities listed in this table is c/o Adial Pharmaceuticals, Inc., 1180 Seminole Trail, Suite 495, Charlottesville, Virginia 22901.
| | | Number of shares
(pro forma) | | | Percentage of
shares | |
| Name and address of beneficial owner | | beneficially
owned | | | beneficially
owned | |
| Directors and named executive officers | | | | | | |
| William B. Stilley, III (Chief Executive Officer, President and Director)(1) | | | 1,830,752 | | | | 10.07 | % |
| Joseph Truluck (Chief Operating Officer and Chief Financial Officer)(2) | | | 372,448 | | | | 2.12 | % |
| J. Kermit Anderson (Director)(3) | | | 33,913 | | | | * | |
| Robertson H. Gilliland, MBA (Director)(4) | | | 33,913 | | | | * | |
| Bankole Johnson, DSc, MD (Chief Medical Officer)(5) | | | 1,278,143 | | | | 7.23 | % |
| James W. Newman, Jr. (Director)(6) | | | 767,562 | | | | 4.33 | % |
| Kevin Schuyler, CFA (Director)(7) | | | 1,477,600 | | | | 8.08 | % |
| Tony Goodman (Director)(8) | | | 55,248 | | | | * | |
| | | | | | | | | |
| All current executive officers and directors as a group (8 persons) | | | 5,849,579 | | | | 28.61 | % |
| (1) | Includes (i) 583,796 shares of common stock, a warrant to acquire 10,829 shares of our common stock having an exercise price of $.0054 per share, a warrant to acquire 36,800 shares of our common having an exercise price of $5.00 per share, a warrant to acquire 5,452 shares of our common stock having an exercise price of $7.63 per share, a warrant to acquire 205,827 shares of our common stock having an exercise price of $6.25 per share; (ii) 333,250 shares of common stock and a warrant to acquire 9,824 shares of our common stock having an exercise price of $7.63 per share owned by Mr. Stilley and his wife Anne T. Stilley. Does not include (x) 5,580 shares of our common stock owned by the Meredith A. Stilley Trust dtd 11/23/2010; (y) 5,580 shares of our common stock owned by the Morgan J. Stilley Trust dtd 11/23/2010; and (z) 5,580 shares of our common stock owned by the Blair E. Stilley Trust dtd 11/23/2010. The trusts are for the benefit of Mr. Stilley’s children and Mr. Stilley is not the trustee. Mr. Stilley disclaims beneficial ownership of these shares except to the extent of any pecuniary interest he may have in such shares. The number of shares reported for Mr. Stilley represents the number of shares he and the trusts received in connection with the corporate conversion/reincorporation and subsequent stock issuances. Includes 644,974 shares of common stock which will have been vested within 60 days of March 22, 2021, which shares were part of total option grants to purchase 1,267,474 shares of our common stock. Of the shares of common stock listed above, 201,109 held by Mr. Stilley and his wife Anne T. Stilley that were issued to them in connection with the acquisition of Purnovate, LLC are subject to a lock-up and are held in escrow as collateral to secure certain of our rights in connection with the acquisition agreement until the earlier of two (2) year anniversary of the closing of the acquisition or on the termination date of Mr. Stilley’s employment if termination is by us without cause. |
| (2) | Comprised of 107,639 shares of our common stock. The number of shares also includes 5,927 warrants to purchase shares of common stock at an exercise price of $6.25 per share. Includes 258,882 shares of common stock, which will vest within 60 days of March 22, 2021, which shares were part of a total option grant to purchase 535,132 shares of our common stock. |
| (3) | Includes 33,913 shares of common stock which will vest within 60 days of March 22, 2021, which shares were part of total option grants to purchase 105,580 shares of our common stock. |
| (4) | Includes 33,913 shares of common stock which will vest within 60 days of March 22, 2021, which shares were part of total option grants to purchase 105,580 shares of our common stock. |
| (5) | Includes (i) 698,336 shares of our common stock owned by En Fideicomiso De Mi Vida 11/23/2010 (Trust); (ii) 93,000 shares of our common stock owned by En Fidecomiso de Todos Mis Suenos Grantor Retained Annuity Trust dated June 27, 2017; (iii) 1,055 shares of our common stock, a warrant to purchase 3,275 shares of our common stock having an exercise price of $7.63, warrants to purchase 189,714 shares of our common stock having an exercise price of $6.25, a warrant to purchase 17,600 shares of our common stock having an exercise price of $5.00 per share, all owned directly by Bankole A. Johnson; (iv) 22,320 shares of our common stock owned by En Fideicomiso De Mis Suenos 11/23/2010 (Trust); (v) 10,000 shares of our common stock owned by De Mi Amor 11/23/2010 (Trust); (vi) an aggregate of 9,300 shares of our common stock owned by Efunbowale Johnson, Ade Johnson, Lola Johnson, Lina Tiouririne, and Aida Tiouririne from whom Dr. Johnson has an voting proxy, (vi) 40,463 shares of our common stock owned by Medico -Trans Company, LLC. Medico -Trans Company, LCC is controlled by Bankole Johnson. Dr. Johnson is the Trustee of each Trust. Includes 193,080 shares of common stock which will have been vested within 60 days of March 22, 2021, which shares were part of total option grants to purchase 255,580 shares of our common stock. Dr. Johnson executed a guaranty, dated December 12, 2019, of PEPCO’s performance under the Master Services Agreement, dated July 5, 2019, and statement of work (the “Guaranty”), together with a pledge and security agreement, dated December 12, 2019 (the “Pledge and Security Agreement”), to secure the Guaranty with 600,000 shares our common stock beneficially owned by him and a lock-up agreement, dated December 12, 2019, and amended August 19, 2020, to allow the private sale of 350,000 shares of Dr. Johnson’s common stock, (the “Lock-Up”), pursuant to which he agreed not to transfer or dispose of, directly or indirectly, any shares of our common stock, as currently owned by him, until after April 1, 2021. |
| (6) | Includes (i) 152,963 shares of common stock, a warrant to purchase 5,415 shares of our common stock having an exercise price of $.0054 per share, a warrant to purchase 4,974 shares of our common stock having an exercise price of $7.63 per share, a warrant to acquire 205,715 shares of our common stock having an exercise price of $6.25 per share, and a warrant to acquire 92,000 shares of common stock having an exercise price of $5.00 per share, all owned by Virga Ventures, LLC; (ii) 41,160 shares of our common stock a warrant to acquire 29,931 shares of our common stock at an exercise price of $6.25 per share and a warrant to acquire 2,372 shares of our common stock having an exercise price of $7.63 per share, all owned by Newman GST Trust FBO James W. Newman Jr; (iii) 45,221 shares of our common stock, a warrant to acquire 1,186 shares of our common stock having an exercise price of $7.63 per share and a warrant to acquire 45,178 shares of our common stock having an exercise price of $6.25 per share, and a warrant to acquire 20,000 shares of our common stock having an exercise price of $5.00 per share, all owned by Ivy Cottage Group, LLC.; (iv) 24,475 shares of our common stock, a warrant to acquire 2,707 shares of our common stock having an exercise price of $.0054 per share, a warrant to acquire 708 shares of our common stock having an exercise price of $7.63 per share, all owned by Rountop Limited Partnership, LLP; (v) 24,644 shares of common stock and a warrant to acquire 10,000 shares of common stock having an exercise price of $6.25 per share held in a Roth IRA for the benefit of Mr. Newman; (vi) 10,000 shares of common stock and a warrant to acquire 10,000 shares of common stock having an exercise price of $6.25 per share, all owned directly by Mr. Newman, and (vii) 5,000 shares of common stock owned by Courtney Newman, daughter of Mr. Newman. Mr. Newman is the sole member of Virga Ventures, LLC, the general partner of Ivy Cottage Group, LLC and Rountop Limited Partnership, LLP, and Trustee of the Newman GST Trust. Includes 33,913 shares of common stock which will vest within 60 days of March 22, 2021, which shares were part of total option grants to purchase 105,580 shares of our common stock. Of the shares of our common stock listed above, 2,544 held by Virga Ventures, LLC and 1,187 held by Rountop Limited Partnership, LLP that were issued to them in connection with the acquisition of Purnovate are subject to a lock-up and are held in escrow as collateral to secure certain of our rights in connection with the acquisition agreement until five (5) days after the effective date of a registration statement registering such shares with respect to thirty percent (30%) of such shares and on the one (1) year anniversary of the closing of the acquisition with respect to seventy percent (70%) of such shares. |
| (7) | Includes (i) 312,990 shares of common stock, warrants to acquire 1,010 shares of common stock at an exercise price of $.0054 per share, warrants to acquire 351,661 shares of our common stock having an exercise price of $6.25 per share issued upon consummation of our initial public offering, warrant to acquire 8,649 shares common stock at an exercise price of $7.63 per share, and a warrant to acquire 89,600 shares of our common stock having an exercise price of $5.00 per share, all owned directly by Mr. Schuyler (ii) 3,042 shares of our common stock and a warrant to acquire 1,963 shares of our common stock at an exercise price of $.0054 per share, and a warrant to acquire 1,172 shares of common stock at exercise price of $7.63, owned by Carolyn M. Schuyler, his wife, and (iii) 144,200 shares of common stock, warrants to acquire 336,800 shares of common stock having an exercise price of $6.25 per share, and a warrant to acquire 192,600 shares of our common stock having an exercise price of $5.00 per share, all owned directly by MVA 151 Investors, LLC. MVA 151 Investors, LLC is an entity under Mr. Schuyler’s control. Includes 33,913 shares of common stock which will vest within 60 days of March 22, 2021, which shares were part of total option grants to purchase 105,580 shares of our common stock. |
| (8) | Includes 8,755 shares of our common stock our common stock and a warrant to acquire 7,000 shares of our common stock having an exercise price of price of $6.25 per share issued upon consummation of our initial public offering. Mr. Goodman has also been granted an option to purchase 111,160 shares of our common stock, of which 39,493 are vested and exercisable within 60 days of March 22, 2021. |
DESCRIPTION OF OUR SECURITIES
The following description of our capital stock and the provisions of our certificate of incorporation and our bylaws are summaries and are qualified by reference to the certificate of incorporation and the bylaws. We have filed copies of these documents with the SEC as exhibits to our registration statement of which this prospectus forms a part.
General
As of the date of this prospectus, our authorized capital stock consists of 50,000,000 shares of common stock, par value $0.001 per share, and 5,000,000 shares of preferred stock, par value $0.001 per share.
Common Stock
Common stock outstanding. As of March 22, 2021, there were 17,269,877 shares of our common stock outstanding.
Voting rights. The holders of common stock are entitled to one vote per share on all matters to be voted upon by the stockholders, except on matters relating solely to terms of preferred stock. Stockholders do not have cumulative voting rights.
Dividend rights. Subject to preferences that may be applicable to any outstanding preferred stock, the holders of common stock are entitled to receive ratably such dividends, if any, as may be declared from time to time by the board of directors out of funds legally available therefor. See “Dividend Policy.”
Rights upon liquidation. In the event of our liquidation, dissolution or winding up, the holders of common stock are entitled to share ratably in all assets remaining after payment of liabilities, subject to prior distribution rights of preferred stock, if any, then outstanding.
Other rights. The holders of our common stock have no preemptive or conversion rights or other subscription rights. There are no redemption or sinking fund provisions applicable to our common stock.
Preferred Stock
There are no shares of our preferred stock outstanding as of the date of this prospectus.
Our board of directors has the authority to issue preferred stock in one or more classes or series and to fix the designations, powers, preferences and rights, and the qualifications, limitations or restrictions thereof, including dividend rights, conversion right, voting rights, terms of redemption, liquidation preferences and the number of shares constituting any class or series, without further vote or action by the stockholders. Although we have no present plans to issue any other shares of preferred stock, the issuance of shares of preferred stock, or the issuance of rights to purchase such shares, could decrease the amount of earnings and assets available for distribution to the holders of common stock, could adversely affect the rights and powers, including voting rights, of the common stock, and could have the effect of delaying, deterring or preventing a change of control of us or an unsolicited acquisition proposal. To date, no preferred stock has been issued.
Warrants
At the date of this prospectus, we had outstanding warrants to purchase 7,957,225 shares of common stock at exercise prices ranging from $0.005 to $7.634 (with a weighted average exercise price of $4.83) and expiration dates from July 31, 2023 to December 31, 2031.
Warrants Issued in our Initial Public Offering
On July 31, 2018, we consummated our initial public offering (“IPO”) and issued an aggregate of 1,464,000 units, each unit consisting of one share of our common stock and one warrant to purchase one share of our common stock, at a public offering price of $5.00 per unit, before underwriting discounts and expenses. The warrants issued in the IPO are exercisable at any time after their original issuance and at any time up to the date that is five years after their original issuance. The warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice and, at any time a registration statement registering the issuance of the shares of common stock underlying the warrants under the Securities Act is effective and available for the issuance of such shares, or an exemption from registration under the Securities Act is available for the issuance of such shares, by payment in full in immediately available funds for the number of shares of common stock purchased upon such exercise. If a registration statement registering the issuance of the shares of common stock underlying the warrants under the Securities Act is not effective or available and an exemption from registration under the Securities Act is not available for the issuance of such shares, the holder may, in its sole discretion, elect to exercise the warrant through a cashless exercise, in which case the holder would receive upon such exercise the net number of shares of common stock determined according to the formula set forth in the warrant. No fractional shares of common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, we will pay the holder an amount in cash equal to the fractional amount multiplied by the exercise price. A holder will not have the right to exercise any portion of the warrant if the holder (together with its affiliates) would beneficially own in excess of 4.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the warrants. However, any holder may increase or decrease such percentage to any other percentage not in excess of 9.99%, provided that any increase in such percentage shall not be effective until 61 days following notice from the holder to us. The exercise price per whole share of our common stock purchasable upon exercise of the warrants is $6.25 per share (based on the initial public offering price of $5.00 per unit) or 125 % of public offering price of the common stock. The exercise price is subject to appropriate adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting our common stock and also upon any distributions of assets, including cash, stock or other property to our stockholders. Subject to applicable laws, the warrants may be offered for sale, sold, transferred or assigned without our consent.
The warrants issued in the IPO are trading on the Nasdaq Capital Market under the symbol “ADILW.” The warrants were issued in registered form under a warrant agent agreement between VStock Transfer, LLC, as warrant agent, and us. The warrants shall initially be represented only by one or more global warrants deposited with the warrant agent, as custodian on behalf of The Depository Trust Company (DTC) and registered in the name of Cede & Co., a nominee of DTC, or as otherwise directed by DTC. In the event of a fundamental transaction, as described in the warrants and generally including any reorganization, recapitalization or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of more than 50% of our outstanding common stock, or any person or group becoming the beneficial owner of 50% of the voting power represented by our outstanding common stock, the holders of the warrants will be entitled to receive upon exercise of the warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the warrants immediately prior to such fundamental transaction.
Representative’s Warrants
The representative of the underwriters in the IPO were issued warrants to purchase up to a total of 58,560 shares of common stock (4% of the shares of common stock sold in the IPO, excluding the over-allotment). The warrants are exercisable at any time, and from time to time, in whole or in part, during the four-year period commencing one year from the effective date of the IPO, which period shall not extend further than five years from the effective date of the IPO in compliance with FINRA Rules. The warrants are exercisable at a per share price equal to $6.25 per share, or 125% of the public offering price per unit in the IPO (based on the initial offering price of $5.00 per unit). The representative (or permitted assignees under FINRA Rules) will not sell, transfer, assign, pledge, or hypothecate these warrants or the securities underlying these warrants, nor will they engage in any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of the warrants or the underlying securities for a period of 180 days from the effective date of the IPO. In addition, the warrants provide for registration rights upon request, in certain cases. In addition, the warrants provide for registration rights upon request, in certain cases. The demand registration right provided will not be greater than five years from the effective date of the offering in compliance with FINRA Rules. The piggyback registration right provided will not be greater than seven years from the effective date of the offering in compliance with FINRA Rules. We will bear all fees and expenses attendant to registering the securities issuable on exercise of the warrants other than underwriting commissions incurred and payable by the holders. The exercise price and number of shares issuable upon exercise of the warrants may be adjusted in certain circumstances including in the event of a stock dividend, extraordinary cash dividend or our recapitalization, reorganization, merger or consolidation. However, the warrant exercise price or underlying shares will not be adjusted for issuances of shares of common stock at a price below the warrant exercise price.
As of the date of this prospectus, we had outstanding warrants to purchase an aggregate of 1,575,112 shares of our common stock, which warrants were issued in the IPO, calculated as follows: (i) 1,516,552 shares of our common stock issuable upon exercise of warrants issued to investors and (ii) 58,560 shares of our common stock issuable upon exercise of warrants issued to the representative of the underwriters.
February 2019 Follow-on Offering
On February 22, 2019, we closed a firm commitment underwritten public offering pursuant to which we issued and sold 2,845,000 shares of our common stock together with a number of warrants to purchase 2,133,750 shares of our common stock. The combined public offering price was $3.25 per share of common stock and accompanying warrant. The warrants are exercisable upon issuance at a price of $4.0625 per share of common stock, subject to adjustment in certain circumstances, and will expire on February 26, 2024. The warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice and, at any time a registration statement registering the issuance of the shares of common stock underlying the warrants under the Securities Act is effective and available for the issuance of such shares, or an exemption from registration under the Securities Act is available for the issuance of such shares, by payment in full in immediately available funds for the number of shares of common stock purchased upon such exercise. If a registration statement registering the issuance of the shares of common stock underlying the warrants under the Securities Act is not effective or available and an exemption from registration under the Securities Act is not available for the issuance of such shares, the holder may, in its sole discretion, elect to exercise the warrant through a cashless exercise, in which case the holder would receive upon such exercise the net number of shares of common stock determined according to the formula set forth in the warrant. Except as otherwise provided in the warrants or by virtue of such holder’s ownership of shares of common stock, the holder of a warrant does not have the rights or privileges of a holder of our common stock, including any voting rights, until the holder exercises the warrant. No fractional shares of common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, at our election, we will pay the holder an amount in cash equal to the fractional amount multiplied by the fair market value of any such fractional shares or round up to the next whole share. The warrants also provide that in the event of a fundamental transaction we are required to cause any successor entity to assume its obligations under the warrants. In addition, the holder of the warrant will be entitled to receive upon exercise of the warrant the kind and amount of securities, cash or property that the holder would have received had the holder exercised the warrant immediately prior to such fundamental transaction. A holder will not have the right to exercise any portion of the warrant if the holder (together with its affiliates) would beneficially own in excess of 4.99% (or, upon election of the holder, 9.99%) of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the warrants. However, any holder may increase or decrease such percentage, provided that any increase will not be effective until the 61st day after such election.
Anti-Takeover Effects of Delaware Law
The provisions of Delaware law, our certificate of incorporation and our bylaws described below may have the effect of delaying, deferring or discouraging another party from acquiring control of us.
Section 203 of the Delaware General Corporation Law
We are subject to Section 203 of the Delaware General Corporation Law, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years after the date that such stockholder became an interested stockholder, with the following exceptions:
| | ● | before such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; |
| | | |
| | | upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the interested stockholder) those shares owned (i) by persons who are directors and also officers and (ii) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
| | ● | on or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of the stockholders, and not by written consent, by the affirmative vote of at least sixty-six and two-thirds percent (66 2/3%) of the outstanding voting stock that is not owned by the interested stockholder. |
In general, Section 203 defines business combination to include the following:
| | ● | any merger or consolidation involving the corporation and the interested stockholder; |
| | | |
| | ● | any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder; |
| | | |
| | ● | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; |
| | | |
| | ● | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or |
| | | |
| | ● | the receipt by the interested stockholder of the benefit of any loss, advances, guarantees, pledges or other financial benefits by or through the corporation. |
Certificate of Incorporation and Bylaws
Our certificate of incorporation and bylaws provide that:
| | ● | our board of directors is divided into three classes, one class of which is elected each year by our stockholders with the directors in each class to serve for a three-year term; |
| | | |
| | ● | the authorized number of directors can be changed only by resolution of our board of directors; |
| | | |
| | ● | directors may be removed only by the affirmative vote of the holders of at least 60% of our voting stock, whether for cause or without cause; |
| | | |
| | ● | our bylaws may be amended or repealed by our board of directors or by the affirmative vote of sixty-six and two-thirds percent (66 2/3%) of our stockholders; |
| | | |
| | ● | stockholders may not call special meetings of the stockholders or fill vacancies on the board of directors; |
| | | |
| | ● | our board of directors will be authorized to issue, without stockholder approval, preferred stock, the rights of which will be determined at the discretion of the board of directors and that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that our board of directors does not approve; |
| | | |
| | ● | our stockholders do not have cumulative voting rights, and therefore our stockholders holding a majority of the shares of common stock outstanding will be able to elect all of our directors; and |
| | | |
| | ● | our stockholders must comply with advance notice provisions to bring business before or nominate directors for election at a stockholder meeting. |
Board Classification
Our board of directors is divided into three classes, one class of which is elected each year by our stockholders. The directors in each class will serve for a three-year term. For more information on the classified board, see “Management—Board of Directors and Executive Officers.” The classification of our board of directors and the limitations on the ability of our stockholders to remove directors could make it more difficult for a third-party to acquire, or discourage a third-party from seeking to acquire, control of us.
Potential Effects of Authorized but Unissued Stock
We have shares of common stock and preferred stock available for future issuance without stockholder approval. We may utilize these additional shares for a variety of corporate purposes, including future public offerings to raise additional capital, to facilitate corporate acquisitions or payment as a dividend on the capital stock.
The existence of unissued and unreserved common stock and preferred stock may enable our board of directors to issue shares to persons friendly to current management or to issue preferred stock with terms that could render more difficult or discourage a third-party attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise, thereby protecting the continuity of our management. In addition, the board of directors has the discretion to determine designations, rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences of each series of preferred stock, all to the fullest extent permissible under the Delaware General Corporation Law and subject to any limitations set forth in our certificate of incorporation. The purpose of authorizing the board of directors to issue preferred stock and to determine the rights and preferences applicable to such preferred stock is to eliminate delays associated with a stockholder vote on specific issuances. The issuance of preferred stock, while providing desirable flexibility in connection with possible financings, acquisitions and other corporate purposes, could have the effect of making it more difficult for a third party to acquire, or could discourage a third party from acquiring, a majority of our outstanding voting stock.
Limitations of Director Liability and Indemnification of Directors, Officers and Employees
Our certificate of incorporation limits the liability of directors to the maximum extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except for liability for any:
| | ● | breach of their duty of loyalty to us or our stockholders; |
| | | |
| | ● | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| | | |
| | ● | unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the Delaware General Corporation Law; or |
| | | |
| | ● | transaction from which the directors derived an improper personal benefit. |
These limitations of liability do not apply to liabilities arising under the federal or state securities laws and do not affect the availability of equitable remedies such as injunctive relief or rescission.
Our bylaws provide that we will indemnify our directors and officers to the fullest extent permitted by law, and may indemnify employees and other agents. Our bylaws also provide that we are obligated to advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding.
We have obtained a policy of directors’ and officers’ liability insurance.
We have entered into separate indemnification agreements with our directors and officers. These agreements, among other things, require us to indemnify our directors and officers for any and all expenses (including reasonable attorneys’ fees, retainers, court costs, transcript costs, fees of experts, witness fees, travel expenses, duplicating costs, printing and binding costs, telephone charges, postage, delivery service fees) judgments, fines and amounts paid in settlement actually and reasonably incurred by such directors or officers or on his or her behalf in connection with any action or proceeding arising out of their services as one of our directors or officers, or any of our subsidiaries or any other company or enterprise to which the person provides services at our request provided that such person follows the procedures for determining entitlement to indemnification and advancement of expenses set forth in the indemnification agreement. We believe that these bylaw provisions and indemnification agreements are necessary to attract and retain qualified persons as directors and officers.
The limitation of liability and indemnification provisions in our certificate of incorporation and bylaws may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might provide a benefit to us and our stockholders. Our results of operations and financial condition may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
At present, there is no pending litigation or proceeding involving any of our directors or officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
Requirements for Advance Notification of Stockholder Nominations and Proposals
Our Bylaws establish advance notice procedures with respect to stockholder proposals and nomination of candidates for election as directors.
Limits on Special Meetings
Special meetings of the stockholders may be called at any time only by the board of directors, Chairman or our Chief Executive Officer, subject to the rights of the holders of any series of preferred stock.
Election and Removal of Directors
Directors are elected by a plurality of the votes of shares present in person or represented by proxy at a meeting and entitled to vote generally on the election of directors. Our stockholders may remove directors only with the vote of sixty percent (60%) of the stockholders, whether for cause or without cause. Our board of directors may appoint a director to fill a vacancy, including vacancies created by the expansion of the board of directors. This system of electing and removing directors may discourage a third party from making a tender offer or otherwise attempting to obtain control of us, because it generally makes it more difficult for stockholders to replace a majority of our directors. Our certificate of incorporation and bylaws do not provide for cumulative voting in the election of directors.
Amendments to Our Governing Documents
Generally, the amendment of our certificate of incorporation requires approval by our board of directors and a majority vote of stockholders. Any amendment to our bylaws requires the approval of either a majority of our board of directors or approval of at least sixty-six and two-thirds (66 2/3%) of the votes entitled to be cast by the holders of our outstanding capital stock in elections of our board of directors.
Choice of Forum
Our certificate of incorporation provides that the Court of Chancery of the State of Delaware will be the exclusive forum for:
| | ● | any derivative action or proceeding brought on our behalf; |
| | | |
| | ● | any action asserting a breach of fiduciary duty; any action asserting a claim against us arising pursuant to the Delaware General Corporation Law, our certificate of incorporation or our bylaws; or |
| | | |
| | ● | any action asserting a claim against us that is governed by the internal affairs doctrine. |
The exclusive forum provision does not apply to suits brought to enforce any liability or duty created by the Securities Act or the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. To the extent that any such claims may be based upon federal law claims, Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. Furthermore, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder.
These exclusive-forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, employees, control persons, underwriters, or agents, which may discourage lawsuits against us and our directors, employees, control persons, underwriters, or agents. Additionally, a court could determine that the exclusive forum provision is unenforceable, and our stockholders will not be deemed to have waived our compliance with the federal securities laws and the rules and regulations thereunder. If a court were to find these provisions of our bylaws inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business, financial condition, or results of operations.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is VStock Transfer, LLC. The transfer agent’s address is 18 Lafayette Place, Woodmere, New York 11598.
Listing on the Nasdaq Capital Market
Our common stock is listed on the Nasdaq Capital Market under the symbol “ADIL.” Our warrants issued in connection with our initial public offering in July 2018 are currently listed on the Nasdaq Capital Market under the symbol “ADILW.”
PLAN OF DISTRIBUTION
Issuance of Shares Upon Exercise of Warrants
The prices at which the shares of common stock covered by this prospectus may actually be disposed of may be at fixed prices, at prevailing market prices at the time of sale, at prices related to the prevailing market price, at varying prices determined at the time of sale or at negotiated prices.
Pursuant to the terms of the warrants, the shares of common stock will be distributed to those holders who surrender the warrants and provide payment of the exercise price to us.
Upon receipt of proper notice by any of the holders of the warrants issued that such holder desires to exercise a warrant, we will, within the time allotted by the agreement governing the warrant, issue instructions to our transfer agent to issue to the holder shares of common stock, free of a restrictive legend. Warrant Shares that are held by affiliates will be issued free of legend but will be deemed control securities.
LEGAL MATTERS
The validity of the securities being offered by this prospectus have been passed upon for us by Gracin & Marlow, LLP, New York, New York.
EXPERTS
The financial statements of Adial Pharmaceuticals, Inc. as of December 31, 2020 and 2019 and for each of the years in the two year period ended December 31, 2020 included in this Registration Statement have been so included in reliance on the report of Friedman LLP, an independent registered public accounting firm, (such report includes an explanatory paragraph regarding the Company’s ability to continue as a going concern), given on the authority of said firm as experts in auditing and accounting.
WHERE YOU CAN FIND MORE INFORMATION
We file annual, quarterly and special reports, proxy statements and other information with the SEC. Our public filings are available to the public at the SEC’s website at www.sec.gov.
Our website address is www.adialpharma.com. Through our website, we make available, free of charge, the following documents as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC: our Annual Reports on Form 10-K; our proxy statements for our annual and special stockholder meetings; our Quarterly Reports on Form 10-Q; our Current Reports on Form 8-K; Forms 3, 4 and 5 and Schedules 13G and 13D filed on behalf of our directors and our executive officers; and amendments to those documents. The information contained on, or that may be accessed through, our website is not part of, and is not incorporated into, this prospectus.
This prospectus is part of a registration statement on Form S-1 that we have filed with the SEC under the Securities Act. This prospectus does not contain all of the information in the registration statement. We have omitted certain parts of the registration statement, as permitted by the rules and regulations of the SEC. You may inspect and print copies of the registration statement, including exhibits, at the SEC’s website or our website.
DISCLOSURE OF COMMISSION POSITION ON INDEMNIFICATION
FOR SECURITIES ACT LIABILITIES
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers, and controlling persons, we have been informed that in the opinion of the SEC this indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
ADIAL PHARMACEUTICALS, INC.
FINANCIAL STATEMENTS
Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of Adial Pharmaceuticals, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Adial Pharmaceuticals, Inc. (the “Company”) as of December 31, 2020 and 2019, and the related statements of operations, stockholders’ equity, and cash flows for each of the years in the two-year period ended December 31, 2020, and the related notes (collectively referred to as the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2020 and 2019, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2020, in conformity with accounting principles generally accepted in the United States of America.
The Company’s Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. As described in Note 2, the Company has an accumulated deficit of $31.5 million as of December 31, 2020 and has suffered recurring losses since inception. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that may result from the outcome of these uncertainties.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/ Friedman LLP | |
| | |
| We have served as the Company’s auditor since 2017. |
| East Hanover, New Jersey | |
| March 22, 2021 | |
ADIAL PHARMACEUTICALS, INC.
BALANCE SHEETS
| | | December 31,
2020 | | | December 31,
2019 | |
| ASSETS | | | | | | |
| Current Assets: | | | | | | |
| Cash and cash equivalents | | $ | 4,401,114 | | | $ | 6,777,052 | |
| Prepaid research and development | | | 233,035 | | | | 536,916 | |
| Prepaid expenses and other current assets | | | 501,689 | | | | 359,499 | |
| Total Current Assets | | | 5,135,838 | | | | 7,673,467 | |
| | | | | | | | | |
| Intangible assets, net | | | 5,606 | | | | 6,170 | |
| Advance to seller | | | 350,000 | | | | — | |
| | | | | | | | | |
| Total Assets | | $ | 5,491,444 | | | $ | 7,679,637 | |
| | | | | | | | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | |
| Current Liabilities: | | | | | | | | |
| Accounts payable | | $ | 648,739 | | | $ | 190,204 | |
| Accrued expenses | | | 856,639 | | | | 348,847 | |
| Total Current Liabilities | | | 1,505,378 | | | | 539,051 | |
| | | | | | | | | |
| Commitments and contingencies | | | | | | | | |
| | | | | | | | | |
| Stockholders’ Equity | | | | | | | | |
| Preferred Stock, 5,000,000 shares authorized with a par value of $0.001 per share, 0 shares outstanding at December 31, 2020 and 2019 | | | — | | | | — | |
| Common Stock, 50,000,000 shares authorized with a par value of $0.001 per share, 14,393,100 and 10,368,352 shares issued and outstanding at December 31, 2020 and 2019, respectively | | | 14,393 | | | | 10,368 | |
| Additional paid in capital | | | 35,491,462 | | | | 27,757,017 | |
| Accumulated deficit | | | (31,519,789 | ) | | | (20,626,799 | ) |
| Total Stockholders’ Equity | | | 3,986,066 | | | | 7,140,586 | |
| | | | | | | | | |
| Total Liabilities and Stockholders’ Equity | | $ | 5,491,444 | | | $ | 7,679,637 | |
The accompanying notes are an integral part of these financial statements.
ADIAL PHARMACEUTICALS, INC.
STATEMENTS OF OPERATIONS
| | | For the Years Ended
December 31, | |
| | | 2020 | | | 2019 | |
| Operating Expenses: | | | | | | |
| Research and development | | $ | 5,853,291 | | | $ | 3,965,543 | |
| General and administrative | | | 5,074,694 | | | | 4,279,357 | |
| Total Operating Expenses | | | 10,927,985 | | | | 8,244,900 | |
| | | | | | | | | |
| Loss From Operations | | | (10,927,985 | ) | | | (8,244,900 | ) |
| | | | | | | | | |
| Other Income (Expense) | | | | | | | | |
| Interest income | | | 32,495 | | | | 95,234 | |
| Other income | | | 2,500 | | | | — | |
| Warrant modification expense | | | — | | | | (441,763 | ) |
| Total other income (expense) | | | 34,995 | | | | (346,529 | ) |
| | | | | | | | | |
| Loss Before Provision For Income Taxes | | | (10,892,990 | ) | | | (8,591,429 | ) |
| Benefit from income taxes | | | — | | | | — | |
| | | | | | | | | |
| Net Loss | | $ | (10,892,990 | ) | | $ | (8,591,429 | ) |
| | | | | | | | | |
| Net loss per share, basic and diluted | | $ | (0.87 | ) | | $ | (0.87 | ) |
| | | | | | | | | |
| Weighted average shares, basic and diluted | | | 12,463,127 | | | | 9,852,486 | |
The accompanying notes are an integral part of these financial statements.
ADIAL PHARMACEUTICALS, INC.
STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
FOR THE YEARS ENDED DECEMBER 31, 2020 and 2019
| | | Common Stock | | | Additional
Paid In | | | Accumulated | | | Total
Stockholders’ | |
| | | Shares | | | Amount | | | Capital | | | Deficit | | | Equity | |
| Balance, December 31, 2018 | | | 6,862,499 | | | $ | 6,863 | | | $ | 16,469,818 | | | $ | (12,035,370 | ) | | $ | 4,441,311 | |
| Stock-based compensation | | | — | | | | — | | | | 1,078,573 | | | | — | | | | 1,078,573 | |
| Stock-based compensation - common stock issued for services | | | 234,437 | | | | 234 | | | | 523,511 | | | | — | | | | 523,745 | |
| Warrant modification expense | | | — | | | | — | | | | 441,763 | | | | — | | | | 441,763 | |
| Sale of common stock & warrants | | | 2,845,000 | | | | 2,845 | | | | 9,243,404 | | | | — | | | | 9,246,249 | |
| Offering issuance cost | | | — | | | | — | | | | (1,050,576 | ) | | | — | | | | (1,050,576 | ) |
| Exercise of warrants | | | 426,416 | | | | 426 | | | | 1,050,524 | | | | — | | | | 1,050,950 | |
| Net loss | | | — | | | | — | | | | — | | | | (8,591,429 | ) | | | (8,591,429 | ) |
| Balance, December 31, 2019 | | | 10,368,352 | | | $ | 10,368 | | | $ | 27,757,017 | | | $ | (20,626,799 | ) | | $ | 7,140,586 | |
| Stock-based compensation | | | — | | | | — | | | | 1,420,455 | | | | — | | | | 1,420,455 | |
| Stock-based compensation, common stock issued for services | | | 446,251 | | | | 446 | | | | 710,346 | | | | — | | | | 710,792 | |
| Sale of common stock & warrants | | | 3,177,143 | | | | 3,177 | | | | 5,713,823 | | | | — | | | | 5,717,000 | |
| Offerings issuance costs | | | — | | | | — | | | | (559,785 | ) | | | — | | | | (559,785 | ) |
| Warrant exercises | | | 226,354 | | | | 227 | | | | 449,781 | | | | — | | | | 450,008 | |
| Issuance of commitment shares | | | 175,000 | | | | 175 | | | | (175 | ) | | | — | | | | — | |
| Net loss | | | — | | | | — | | | | — | | | | (10,892,990 | ) | | | (10,892,990 | ) |
| Balance, December 31, 2020 | | | 14,393,100 | | | $ | 14,393 | | | $ | 35,491,462 | | | $ | (31,519,789 | ) | | $ | 3,986,066 | |
The accompanying notes are an integral part of these financial statements.
ADIAL PHARMACEUTICALS, INC.
STATEMENTS OF CASH FLOWS
| | | For the Years Ended
December 31, | |
| | | 2020 | | | 2019 | |
| CASH FLOWS FROM OPERATING ACTIVITIES: | | | | | | |
| Net loss | | $ | (10,892,990 | ) | | $ | (8,591,429 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | |
| Stock-based compensation | | | 2,014,246 | | | | 1,602,318 | |
| Non-cash warrant modification expense | | | — | | | | 441,763 | |
| Amortization of intangible assets | | | 564 | | | | 565 | |
| Changes in operating assets and liabilities: | | | | | | | | |
| Prepaid research and development | | | 303,881 | | | | (30,956 | ) |
| Prepaid expenses and other current assets | | | (142,190 | ) | | | (41,952 | ) |
| Accounts payable | | | 458,535 | | | | 90,533 | |
| Accrued expenses | | | 624,793 | | | | 190,544 | |
| Net cash used in operating activities | | | (7,633,161 | ) | | | (6,338,614 | ) |
| | | | | | | | | |
| CASH FLOWS FROM INVESTING ACTIVITIES: | | | | | | | | |
| Advance to seller | | | (350,000 | ) | | | — | |
| Net cash used in investing activities | | | (350,000 | ) | | | — | |
| | | | | | | | | |
| CASH FLOWS FROM FINANCING ACTIVITIES: | | | | | | | | |
| Net proceeds from equity offerings | | | 5,157,215 | | | | 8,195,673 | |
| Proceeds from warrant exercises | | | 450,008 | | | | 1,050,950 | |
| Net cash provided by financing activities | | | 5,607,223 | | | | 9,246,623 | |
| | | | | | | | | |
| NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS | | | (2,375,938 | ) | | | 2,908,009 | |
| | | | | | | | | |
| CASH AND CASH EQUIVALENTS-BEGINNING OF YEAR | | | 6,777,052 | | | | 3,869,043 | |
| | | | | | | | | |
| CASH AND CASH EQUIVALENTS-END OF YEAR | | $ | 4,401,114 | | | $ | 6,777,052 | |
| | | | | | | | | |
| SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: | | | | | | | | |
| Interest paid | | $ | — | | | $ | — | |
| Income taxes paid | | $ | — | | | $ | — | |
| Reclassification of stock-based comp from accrued expenses | | $ | 117,001 | | | $ | — | |
| Non-cash cost of commitment shares | | $ | 295,750 | | | $ | — | |
The accompanying notes are an integral part of these financial statements.
ADIAL PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS
1 — DESCRIPTION OF BUSINESS
Adial Pharmaceuticals, Inc. (the “Company” or “Adial”) was converted from a limited liability company formed on November 23, 2010 to a corporation and reincorporated in Delaware on October 1, 2017. Adial is presently engaged in the development of medications for the treatment and prevention of addictions and related disorders.
The Company has commenced its first Phase 3 clinical trial of its lead compound AD04 (“AD04”) for the treatment of alcohol use disorder. Both the U.S. Food and Drug Administration (“FDA”) and the European Medicines Authority (“EMA”) have indicated they will accept heavy-drinking-based endpoints as a basis for approval for the treatment of alcohol use disorder rather than the previously required abstinence-based endpoints. Key patents have been issued in the United States, the European Union, and other jurisdictions for which the Company has exclusive license rights. The active ingredient in AD04 is ondansetron, a serotonin-3 antagonist. Due to its mechanism of action, AD04 has the potential to be used for the treatment of other addictive disorders, such as opioid use disorder, obesity, smoking, and other drug addictions.
2 — LIQUIDITY, GOING CONCERN AND OTHER UNCERTAINTIES
These financial statements have been prepared in conformity with generally accepted accounting principles in the United States (“GAAP”), which contemplate continuation of the Company as a going concern. The Company is in a development stage and has incurred losses and negative cash flows from operations each year since inception and has an accumulated deficit of approximately $31.5 million as of December 31, 2020. Based on the current development plans for AD04 and other operating requirements, existing cash and equivalents are not sufficient to fund operations for the next twelve months following the financial statement filing date. These factors raise substantial doubt about the Company’s ability to continue as a going concern.
Due to the COVID-19 pandemic, during the first three quarters of 2020, the Company experienced delays in certain countries in obtaining regulatory approval required to commence the trial in such countries, resulting in significantly slowed trial enrollment (see Other Uncertainties below). Enrollment has since improved, though enrollment in higher cost sites in Scandinavia have recovered more quickly than in lower cost sites in Central and Eastern Europe. The Company presently projects completion of its phase three trial during the first quarter of 2022 with available cash currently projected into the fourth quarter of 2021. As such, cash on hand is not projected to be sufficient to reach database lock.
The Company’s plans include raising additional capital from various potential sources, including equity and/or debt financings, grant funding, and strategic relationships. In addition, the Company has an active equity purchase agreement (See Notes 7 and 10) with Keystone Capital, LLC, which allows the sale of up to $15 million, which amount, if fully accessible, would be sufficient to fund the Company’s operations beyond one year from the filing date of these statements. However, there can be no guarantee that the Company will meet the conditions to use the equity line and without access to this credit line and/or additional funding, which may not be available on acceptable terms or at all, the Company would be required to delay, scale back or eliminate some or all of its research and development programs, which would likely have a material adverse effect on the Company and its financial statements.
The accompanying financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might result from the outcome of the uncertainties described above.
Other Uncertainties
Generally, the industry in which the Company operates subjects the Company to a number of other risks and uncertainties that can affect its operating results and financial condition. Such factors include, but are not limited to: the timing, costs and results of clinical trials and other development activities versus expectations; the ability to obtain regulatory approval to market product candidates; the ability to manufacture products successfully; competition from products sold or being developed by other companies; the price of, and demand for, Company products once approved; the ability to negotiate favorable licensing or other manufacturing and marketing agreements for its products.
The Company also faces the ongoing risk that the coronavirus pandemic may further slow, for an unforeseeable period, the conduct of the Company’s trial. The effects of the ongoing coronavirus pandemic may also increase non-trial costs such as insurance premiums, increase the demand for and cost of capital, increase loss of work time from key personnel, and negatively impact our key clinical trial vendors and supplier of our active pharmaceutical ingredient. The full extent to which the COVID-19 pandemic further impacts the clinical development of AD04, the Company’s suppliers and other commercial partners, will depend on future developments that cannot be predicted.
3 — SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, and disclosure of contingent liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Significant items subject to such estimates and assumptions include the valuation of stock-based compensation, expense recognition and accruals associated with third party providers supporting clinical trials and other research and development, and income tax asset realization.
Basic and Diluted Loss per Share
Basic and diluted loss per share are computed based on the weighted-average outstanding shares of common stock, which are all voting shares. Diluted net loss per share is computed giving effect to all proportional shares of common stock, including stock options and warrants to the extent dilutive. Basic net loss per share was the same as diluted net loss per share for the years ended December 31, 2020 and 2019 as the inclusion of all potential common shares outstanding would have an anti-dilutive effect. “Penny warrants” were not excluded from calculation of outstanding shares for purposes of basic earnings per share.
The total potentially dilutive common shares that were excluded for the years ended December 31, 2020 and 2019 were as follows:
| | | Potentially
Dilutive Common
Shares Outstanding
December 31, | |
| | | 2020 | | | 2019 | |
| Warrants to purchase common shares, less “penny warrants” included in potentially dilutive common shares | | | 8,577,336 | | | | 6,595,631 | |
| Common shares issuable on exercise of options | | | 2,668,866 | | | | 1,661,466 | |
| Total potentially dilutive common shares excluded | | | 11,246,202 | | | | 8,257,097 | |
Cash and Cash Equivalents
The Company considers all highly liquid investments with original maturities of three months or less to be cash equivalents. At times, the Company’s cash balances may exceed the current insured amounts under the Federal Deposit Insurance Corporation. At December 31, 2020, the Company did not exceed FDIC insurance limits but held approximately $4.2 million in non-FDIC insured cash accounts. Included in cash equivalents are equity securities with maturity dates less than ninety days and are carried at fair value. Unrealized gain or loss are included in the interest income and are immaterial to the financial statements. At December 31, 2019, the Company exceeded FDIC insurance limits by approximately $0.4 million and held approximately $6.1 million in non-FDIC insured cash equivalent investments.
Intangible Assets
Intangible assets consist primarily of the trademarks and copyrights. The trademarks and copyrights will be amortized using the straight-line method based on an estimated useful life of 20 years.
Impairment of Long-Lived Assets
The Company’s long-lived assets (consisting of the trademarks) are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the undiscounted future net cash flows expected to be generated by that asset. If the carrying amount of an asset exceeds its estimated future undiscounted cash flows, an impairment charge is recognized by the amount by which the carrying amount of the asset exceeds the fair value of the asset.
Research and Development
Research and development costs are charged to expense as incurred and include direct trial expenses such as fees due to contract research organizations, consultants which support the Company’s research and development endeavors, the acquisition of technology rights without an alternative use, and compensation and benefits of clinical research and development personnel. Certain research and development costs, in particular fees to contract research organizations (“CROs”), are structured with milestone payments due on the occurrence of certain key events. Where such milestone payments are greater than those earned through the provision of such services, the Company recognizes a prepaid asset which is recorded as expense as services are incurred. Where such expenses are greater than total milestone payments made, an accrued expense liability is recognized.
Stock-Based Compensation
The Company accounts for stock-based compensation to employees and non-employees in conformity with the provisions of ASC 718, Compensation - Stock Based Compensation (“ASC 718”). The Company expenses stock-based compensation to employees and non-employees over the requisite service period based on the estimated grant-date fair value of the awards. The Company estimates the fair value of options granted using the Black Scholes Merton model. The Company estimates when and if performance-based awards will be earned. If an award is not considered probable of being earned, no amount of equity-based compensation expense is recognized. If the award is deemed probable of being earned, related equity-based compensation expense is recorded. The fair value of an award ultimately expected to vest is recognized as an expense, net of forfeitures, over the requisite service, which is generally the vesting period of the award.
The Black Scholes Merton model requires the input of certain subjective assumptions and the application of judgment in determining the fair value of the awards. The most significant assumptions and judgments include the expected volatility, risk-free interest rate, the expected dividend yield, and the expected term of the awards.
The key assumptions included in the model are as follows:
| ● | Expected volatility — The expected price volatility based on the historical volatilities of peer group companies as the Company does not have a sufficient trading history. Industry peers consist of several public companies in the bio-tech industry similar in size, stage of life cycle, and capital structure. The Company also blends in historical data on the volatility of its own equity, increasing in proportion as the period of historical data on the Company’s data becomes more representative. |
| ● | Risk-free interest rate — The risk free rate was determined based on yields of U.S. Treasury Bonds of comparable terms. |
| ● | Expected dividend yield — The Company has not previously issued dividends and do not anticipate paying dividends in the foreseeable future. Therefore, we used a dividend rate of zero based on our expectation of additional dividends. |
| ● | Expected term —The expected term of the options was estimated using the simplified method. |
Common shares issued to third parties for services provided are valued based on the fair value of the Company’s common shares as determined by the market closing price of a share of our common stock on the date of the Commitment to make the issuance.
Income Taxes
The Company accounts for income taxes using the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax basis and tax carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
A valuation allowance is established to reduce net deferred tax assets to the amount expected to be realized. The Company recognizes the effect of income tax positions only if those positions are more likely than not of being sustained. Changes in recognition and measurement are reflected in the period in which the change in judgment occurs. Interest and penalties related to unrecognized tax benefits are included in income tax expense. The Company has generally recorded a full valuation allowance for its tax carryforwards, reflecting the judgment of Company management that they are more likely than not to expire unused.
Fair Value of Financial Instruments and Fair Value Measurements
Significant items subject to such estimates and assumptions include the valuation of stock-based compensation, derivative liabilities, accruals associated with third party providers supporting clinical trials, contingent liabilities and income tax liability. Authoritative literature establishes a three-level valuation hierarchy for disclosures of fair value measurements and disclosure. The carrying amounts reported in the balance sheets for current liabilities, convertible notes, Senior Notes, Senior Secured Bridge Notes, and Subordinated Notes are a reasonable estimate of their fair values because of the short period of time between the origination of such instruments and their expected realization and their current market rate of interest. The carrying value of all other financial liabilities at cost approximates fair value.
The three levels of valuation hierarchy are defined as follows:
| ● | Level 1: Observable inputs such as quoted prices in active markets; |
| ● | Level 2: Inputs, other than the quoted prices in active markets, that are observable either directly or indirectly; and |
| ● | Level 3: Unobservable inputs in which there is little or no market data, which require the reporting entity to develop its own assumptions. |
Adoption of Recent Accounting Pronouncements
Fair Value — In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement (Topic 820) Disclosure Framework—Changes to the Disclosure Requirements for Fair Value Measurement (“ASU 2018-13”). ASU 2018-13 amends guidance concerning disclosure of transfers between the Levels 1, 2, and 3 for the fair value hierarchy used to disclose the fair value of financial instruments. ASU 2018-13 also adds additional requirements that reporting entities disclose unrealized gains or losses in the value of financial instruments as a result of changes to recurring fair Level 3 fair value measurements and the range and weighted averages of significant unobservable inputs used to develop fair value measurements. The amendments in ASU 2018-13 are effective for all entities required under existing GAAP to disclose fair value measurements, and is effective for annual periods, and interim periods within those annual periods, beginning after December 15, 2019. ASU 2018-13 was adopted effective January 1, 2020. There was no material effect on the financial statements as a result of the adoption of ASU 2018-13.
4 — ADVANCE TO SELLER – RELATED PARTY
In connection with entry into a December 7, 2020 equity purchase agreement with Purnovate, Inc., an early stage drug development company with a focus on purine chemistry and successor to Purnovate, LLC (henceforth “Purnovate”), the Company loaned Purnovate $350,000 to continue its research and development efforts during the due diligence period specified in the agreement, and issued a 3.5% promissory note due December 7, 2021. In the event the acquisition were terminated, the bridge note would become convertible, at the option of the Company, into equity of Purnovate at a conversion price of $1.50. The proceeds of the Bridge Note were to be used by Purnovate to fund ongoing research and development, working capital, and other general corporate purposes. See Notes 6 and 10.
5 — ACCRUED EXPENSES
Accrued expenses consist of the following:
| | | December 31,
2020 | | | December 31,
2019 | |
| Clinical research organization services and expenses | | $ | 470,991 | | | $ | 16,877 | |
| Employee compensation | | | 322,437 | | | | 263,914 | |
| Minimum license royalties | | | 40,000 | | | | – | |
| Manufacturing expenses | | | 17,060 | | | | – | |
| Legal and consulting services | | | 6,151 | | | | 68,056 | |
| Total accrued expenses | | $ | 856,639 | | | $ | 348,847 | |
6 — RELATED PARTY TRANSACTIONS
In January 2011, the Company entered into an exclusive, worldwide license agreement with The University of Virginia Patent Foundation d/b/a the University of Virginia Licensing and Ventures Group (the “UVA LVG”) for rights to make, use or sell licensed products in the United States based upon patents and patent applications made and held by UVA LVG (the “UVA LVG License”). The Company is required to pay compensation to the UVA LVG, as described Note 9. A certain percentage of these payments by the Company to the UVA LVG may then be distributed to the Company’s former Chairman of the Board who currently serves as the Company’s Chief Medical Officer in his capacity as inventor of the patents by the UVA LVG in accordance with their policies at the time.
On September 21, 2020, the Company concluded a private placement of 357,143 unregistered shares of common stock and above market price of $1.40 per share with Bespoke Growth Partners, Inc. (“Bespoke”). Bespoke is controlled by a consultant who serves as the Company’s Chief Development Officer. Net proceeds of the offering was $500,000.
On December 7, 2020, the Company entered into an Equity Purchase Agreement with Purnovate, LLC to purchase all of the outstanding membership interests of Purnovate from the members of Purnovate (the “Members”), such that after the acquisition, Purnovate would be a wholly owned subsidiary of Adial. Purnovate is a drug development company with a platform focused on developing drug candidates for non-opioid pain reduction and other diseases and disorders potentially targeted with adenosine analogs that are selective, potent, stable, and soluble. Adial’s Chief Executive Officer and board member, William B. Stilley, and another Adial board member were, directly or indirectly, members of Purnovate. Messrs. Stilley and Newman agreed to sell their membership interests on the same terms as the other Members, except that Mr. Stilley is subject to a two (2) year lock up with respect to the sale and transfer of the stock consideration that he receives so long as his employment has not been terminated by the Company without cause prior to the end of such period. Mr. Stilley owned approximately 28.7% of the membership interest of Purnovate and Mr. Newman controlled two entities that, together, own less than 1% of the membership interests of Purnovate.
As a result of the foregoing, the Company formed a Special Committee of independent members of its Board of Directors to review and negotiate the acquisition terms. On January 26, 2021 the acquisition was consummated, and Messrs. Stilley and Newman sold all of their membership interests in Purnovate to the Company (see Note 10).
See Note 9 for related party vendor, consulting, and lease agreements and Note 4 for Advance to Seller.
7 — SHAREHOLDERS’ EQUITY
Common Stock Issuances
On January 22, 2019, the Company issued 250,000 unregistered shares of common stock upon the exercise of a warrant to purchase 300,000 shares of common stock at an exercise price of $3.75 per share for a cash payment of $468,750 and the cashless exercise of the remaining warrant.
On January 31, 2019, the Company issued 22,311 unregistered shares of common stock upon the full cashless exercise of a warrant to purchase 65,130 shares of common stock at an exercise price of $4.99 per share.
On February 22, 2019, the Company concluded a follow-on offering of 2,475,000 shares of common stock and warrants to purchase 1,856,250 shares of common stock at an exercise price of $4.0625 per share. The shares of common stock and accompanying warrants were sold to the public at a price of $3.25 per share and warrant. The underwriters were granted an over-allotment option to purchase up to 371,250 shares of common stock and warrants to purchase 278,437 shares of common stock at a price of $3.25 per share of common stock and warrant. The underwriters partially exercised their over-allotment option by purchasing 370,000 shares of common stock and warrants to purchase 277,500 shares common stock. Gross proceeds of the offering, totaled $9,246,249, which after offering expenses, resulted in net proceeds of $8,195,673.
On June 11, 2020, the Company concluded a registered direct offering of 2,820,000 shares of common stock and in a concurrent private placement the sale of warrants to purchase 2,115,000 shares of common stock at an exercise price of $2.00 per share. The shares of common stock and accompanying warrants were sold directly to the buyers at a combined at-the-market price of $1.85 for a share and three quarters warrant. Gross proceeds of the offering, totaled $5,217,000, which after offering expenses, resulted in net proceeds of $4,657,215.
On July 31, 2020, warrants to purchase 1,354 shares at an exercise price of $0.005 per share were exercised for a cash payment of $8.
On September 21, 2020, the Company concluded a private placement of 357,143 unregistered shares of common stock and above market price of $1.40 per share. Gross proceeds of the offering, totaled $500,000, with no material expenses.
On September 29, 2020, previously registered warrants to purchase 225,000 shares at an exercise fee of $2.00 per share were exercised for a total of $450,000.
On November 18, 2020, the Company issue 175,000 shares of common stock at a cost of $295,750 to Keystone Capital, LLC as commitment shares due on execution of an equity purchase agreement.
During the year ended December 31, 2020, the Company issued 446,251 shares of common stock and 91,705 warrants to purchase shares of common stock to consultants for services rendered and to employees at a total cost for the period of $710,792.
2017 Equity Incentive Plan
On October 9, 2017, the Company adopted the Adial Pharmaceuticals, Inc. 2017 Equity Incentive Plan (the “2017 Equity Incentive Plan”); which became effective on July 31, 2018. Initially, the aggregate number of shares of our common stock that may be issued pursuant to stock awards under the 2017 Equity Incentive Plan was 1,750,000 shares. On September 1, 2020, by a vote of the shareholders, the number of shares issuable under the 2017 Equity Incentive Plan was increased to 5,500,000. At December 31, 2020, the Company had issued 799,438 shares of common stock and had outstanding 2,529,180 options to purchase shares of our common stock. At December 31, 2020, a total of 2,171,382 shares remain issuance under the 2017 Equity Incentive Plan.
Stock Options
The following table provides the stock option activity for the year ended December 31, 2020:
| | | Total
Options
Outstanding | | | Weighted
Average
Remaining
Term
(Years) | | | Weighted
Average
Exercise
Price | | | Weighted
Average
Fair Value
at Issue | |
| Outstanding December 31, 2018 | | | 243,182 | | | | 8.93 | | | $ | 4.88 | | | $ | 4.09 | |
| Issued | | | 1,452,880 | | | | | | | | 3.19 | | | | 2.21 | |
| Cancelled | | | (34,596 | ) | | | | | | | 5.70 | | | | 4.23 | |
| Outstanding December 31, 2019 | | | 1,661,466 | | | | | | | | 3.38 | | | | 2.38 | |
| Issued | | | 1,255,000 | | | | | | | | 1.44 | | | | 1.13 | |
| Cancelled | | | (247,600 | ) | | | | | | | 3.27 | | | | 2.04 | |
| Outstanding December 31, 2020 | | | 2,668,866 | | | | 8.09 | | | | 2.48 | | | | 1.78 | |
| Outstanding December 31, 2020, vested and exercisable | | | 1,242,831 | | | | 7.72 | | | $ | 2.94 | | | $ | 2.13 | |
At December 31, 2020, the intrinsic value totals of the outstanding options were $339,520.
The Company used the Black Scholes valuation model to determine the fair value of the options issued, using the following key assumptions for the years ended December 31, 2020 and 2019:
| | | 2020 | | | 2019 | |
| Fair Value per Share | | $ | 1.36-1.48 | | | $ | 1.45-3.39 | |
| Expected Term | | | 5.75 years | | | | 1.46-5.75 years | |
| Expected Dividend | | | – | | | | – | |
| Expected Volatility | | | 102.39-103.83 | % | | | 97.37-101.09 | % |
| Risk free rate | | | 0.49-0.72 | % | | | 2.32-2.51 | % |
During the year ended December 31, 2020, 1,225,000 options to purchase shares of common stock were granted at a fair value of $1,420,505, an approximate weighted average fair value of $1.13 per option, to be amortized over a service a weighted average period of three years. As of December 31, 2020, $2,344,035 in unrecognized compensation expense will be recognized over a weighted average remaining service period of 2.38 years.
The components of stock-based compensation expense included in the Company’s Statements of Operations for the years ended December 31, 2020 and 2019 are as follows:
| | | Year ended December 31, | |
| | | 2020 | | | 2019 | |
| Research and development options expense | | | 252,173 | | | | 355,229 | |
| Total research and development expenses | | | 252,173 | | | | 355,229 | |
| General and administrative options expense | | | 1,168,281 | | | | 723,344 | |
| Stock and warrants issued to consultants and employees | | | 710,792 | | | | 523,745 | |
| Total general and administrative expenses | | | 1,879,073 | | | | 1,247,089 | |
| Total stock-based compensation expense | | $ | 2,131,246 | | | $ | 1,602,318 | |
Stock Warrants
The following table provides the activity in warrants for the respective periods.
| | | Total
Warrants | | | Weighted
Average
Remaining
Term
(Years) | | | Weighted
Average
Exercise
Price | | | Average
Intrinsic
Value | |
| Outstanding December 31, 2018 | | | 5,054,759 | | | | 5.07 | | | $ | 5.72 | | | | 0.61 | |
| Issued | | | 2,113,750 | | | | | | | | 4.06 | | | | | |
| Exercised | | | (519,235 | ) | | | | | | $ | 4.07 | | | | | |
| Outstanding December 31, 2019 | | | 6,669,274 | | | | 4.23 | | | $ | 5.38 | | | | 0.03 | |
| Issued | | | 2,206,705 | | | | | | | | 1.98 | | | | | |
| Exercised | | | (226,354 | ) | | | | | | $ | 1.99 | | | | | |
| Outstanding December 31, 2020 | | | 8,649,625 | | | | 3.46 | | | $ | 4.60 | | | | 0.02 | |
This table includes warrants to purchase 91,705 shares of common stock issued to consultants, with a total fair value of $110,303 at time of issue, calculated using the Black Scholes model assuming an underlying security values ranging from $1.30 to 2.03 and volatility rate of 103.83%, a risk-free rate of 0.49%, and an expected term of 1.5 years.
During the year ended December 31, 2020, 226,354 warrants to purchase shares of common stock were exercised for total proceeds of $450,008.
8 — INCOME TAXES
A reconciliation of the statutory Federal income tax rate and effective rate of the provision for income taxes is as follows:
| | | Year ended December 31, | |
| | | 2020 | | | 2019 | |
| Computed “expected” tax benefit | | $ | (2,287,528 | ) | | | (1,804,200 | ) |
| Increase (reduction) in income taxes resulting from: | | | | | | | | |
| State Tax, net of federal | | | (446,351 | ) | | | (225,900 | ) |
| Stock Compensation | | | 309,838 | | | | 372,190 | |
| Other | | | 188 | | | | 17,572 | |
| Change in the valuation allowance | | | 2,423,853 | | | | 1,640,338 | |
| Total income tax expense/(benefit) | | $ | — | | | | — | |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities recognized for financial reporting, and the amounts recognized for income tax purposes. The significant components of deferred tax assets as of December 31, 2020 and 2019, respectively, are as follows:
Deferred Tax Assets (rounded)
| | | Total | | | Total | | | Deferred Tax Asset | |
| | | 2020 | | | 2019 | | | 2020 | | | 2019 | |
| Net operating loss carry-forward | | | 23,294,000 | | | | 14,120,000 | | | | 5,996,000 | | | | 3,635,000 | |
| Accrued Expenses | | | 243,000 | | | | — | | | | 62,000 | | | | — | |
| Intangible Assets | | | (1,000 | ) | | | (1,000 | ) | | | — | | | | — | |
| Less: valuation allowance | | | (23,536,000 | ) | | | (14,119,000 | ) | | | (6,058,000 | ) | | | (3,635,000 | ) |
| Total | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
The Company has a net operating loss carry-forward for federal and state tax purposes of approximately $23.3 million at December 31, 2020, that is potentially available to offset future taxable income. The 20-year limitation was eliminated for losses generated after January 1, 2018, giving the taxpayer the ability to carry forward losses indefinitely. However, NOL carry forward arising after January 1, 2018, will now be limited to 80 percent of taxable income. In assessing the realizability of the deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income, net operating loss carryback potential and tax planning strategies in making these assessments.
Based on the above criteria, the Company believes that it is more likely than not that the remaining deferred tax assets will not be realized. Accordingly, the Company has recorded a valuation allowance of $6.0M against the net deferred tax asset that is not realizable.
Section 382 of the Internal Revenue Code (“Section 382”) imposes limitations on a corporation’s ability to utilize net operating losses if it experiences an “ownership change.” In general terms, an ownership change may result from transactions increasing the ownership of certain stockholders in the stock of a corporation by more than 50 percentage points over a three-year period. Any unused annual limitation may be carried over to later years, and the amount of the limitation may under certain circumstances be increased by the built-in gains in assets held by us at the time of the change that are recognized in the five-year period after the change.
The Company has not performed a study to assess whether an ownership change for purposes of Section 382 has occurred, or whether there have been multiple ownership changes since the Company’s inception, due to the significant costs and complexities associated with such study. If the Company has experienced a change in control, as defined by Section 382, at any time since its public offering, utilization of net operating loss carryforwards would be subject to an annual limitation under Section 382. Any limitation may result in expiration of a portion of the net operating losses before utilization.
The Company files tax returns as prescribed by the tax laws of the jurisdictions in which they operate. In the normal course of business, the Company is subject to examination by Federal and state jurisdictions where applicable based on the statute of limitations that apply in each jurisdiction. As of December 31, 2020, open years related to the Federal and state jurisdictions are 2019, 2018 & 2017. Since the Company was not a taxable entity prior to reincorporation, examination of returns for years prior to 2017 will not result in changes to tax liability or benefit. The Company has no open tax audits with any taxing authority as of December 31, 2020.
9 — COMMITMENTS AND CONTINGENCIES
License with University of Virginia Patent Foundation
In January 2011, the Company entered into an exclusive, worldwide license agreement with (the “UVA LVG”) for rights to make, use or sell licensed products in the United States based upon the ten separate patents and patent applications made and held by UVA LVG.
As consideration for the rights granted in the UVA LVG License, the Company is obligated to pay UVA LVG yearly license fees and milestone payments, as well as a royalty based on net sales of products covered by the patent-related rights. More specifically, the Company paid UVA LVG a license issue fee and is obligated to pay UVA LVG (i) annual minimum royalties of $40,000 commencing in 2017; (ii) a $20,000 milestone payments upon dosing the first patient under a Phase 3 human clinical trial of a licensed product, $155,000 upon the earlier of the completion of a Phase 3 trial of a licensed product, partnering of a licensed product, or sale of the Company, $275,000 upon acceptance of an NDA by the FDA, and $1,000,000 upon approval for sale of AD04 in the U.S., Europe or Japan; as well as (iii) royalties equal to a 2% and 1% of net sales of licensed products in countries in which a valid patent exists or does not exist, respectively, with royalties paid quarterly. In the event of a sublicense to a third party, the Company is obligated to pay royalties to UVA LVG equal to a percentage of what the Company would have been required to pay to UVA LVG had it sold the products under sublicense ourselves. In addition, the Company is required to pay to UVA LVG 15% of any sublicensing income.
The license agreement may be terminated by UVA LVG upon sixty (60) days written notice if the Company breaches its obligations thereunder, including failing to make any milestone, failure to make required payments, or the failure to exercise diligence to bring licensed products to market. In the event of a termination, the Company will be obligated to pay all amounts that accrued prior to such termination.
The term of the license continues until the expiration, abandonment or invalidation of all licensed patents and patent applications, and following any such expiration, abandonment or invalidation will continue in perpetuity on a royalty-free, fully paid basis.
The Company executed an amendment, dated December 14, 2017, which changed the dates by which the Company, using commercially reasonable efforts, was to achieve the goals of submitting a New Drug Application to the FDA for a licensed product to December 31, 2024 (from December 31, 2023) and commencing commercialization of an FDA approved product by December 31, 2025 (from December 31, 2024). If the Company were to fail to use commercially reasonable effort and fail to meet either goal, the licensor would have the right to terminate the license.
On December 31, 2019, the Company executed a further amendment to the license agreement which, among other things, removed in its entirety the diligence milestone to initiate a Phase 3 clinical trial by December 31, 2019. Furthermore, the Company agreed to pay upon execution of the amendment the diligence milestone payment of $20,000 that had been due upon initiation of a Phase 3 clinical trial. In addition, the Company agreed to use and will continue to use best efforts to dose a first patient with a Licensed Product (as defined in the License Agreement) in a Phase 3 clinical trial on or before March 31, 2020. In March of 2020, the first patient was dosed with AD04 after having joined the Company’s trial, satisfying this term of the license agreement.
During the year ended December 31, 2020 and 2019, the Company recognized $40,000 in minimum license royalty expenses under this agreement.
Clinical Research Organization (CRO)
On October 31, 2018, the Company entered into a master services agreement (“MSA”) with Crown CRO Oy (“Crown”) for contract clinical research and consulting services. The MSA has a term of five years, automatically renewed for two-year periods, unless either party gives written notice of a decision not to renew the agreement three months prior to automatic renewal. The MSA or a service agreement under it may be terminated by the Company, without penalty, on fourteen days written notice for scientific, administrative, or financial reasons, or if the purpose of the study becomes obsolete. In the event that the MSA or Service Order are terminated, Crown’s actual costs up the date of termination will be payable by the Company, but any unrealized milestones would not be owed.
On November 16, 2018, the Company and Crown entered into Service Agreement 1 under the MSA for a 24 week, multi-centered, randomized, double-blind, placebo-controlled, parallel-group, Phase 3 clinical study of the Company’s lead compound, AD04. On June 28, 2019, the Company and Crown Executed a change order to Service Agreement 1 increasing Crown’s fee from $3,614,513 (€2,958,835 converted to dollars at the Euro/US Dollar exchange rate of 1.2216 as of December 31, 2020) to $3,871,122 (€3,168,895) and rescheduling future milestone payments.
On February 1, 2020, the first site initiation visit (“SIV”) of a study site had been completed and the second milestone of $299,496 was paid. On February 27, 2020, the first potential patient for the study had been screened and the third milestone payment of $297,013 was paid. On June 15, 2020, 50% of sites had been initiated and a fourth milestone payment of $302,843 was paid. On October 20, 2020, 100% of sites had been initiated and a fifth milestone $319,310 was paid. On November 10, 2020, 30% of patients had been enrolled and a sixth milestone payment of $319,013 was paid.
At December 31, 2020, the remaining future milestone payments are shown in the table below, converted to dollars from euros at the exchange rate then prevailing.
| Milestone Event | | Percent
Milestone Fees | | | Amount | |
| 60% patients randomized | | | 10 | % | | $ | 329,756 | |
| 100% of patients randomized | | | 10 | % | | $ | 329,756 | |
| 90% of case report form pages monitored | | | 5 | % | | $ | 164,878 | |
| PE analysis | | | 5 | % | | $ | 164,878 | |
| Database is locked | | | 10 | % | | $ | 329,756 | |
During the year ended December 31, 2020, the Company recognized $1,850,495 in direct expenses associated with the Service Agreement 1, classified as R&D expense. Prepaid R&D costs were $214,633 at December 31, 2019, and on December 31, 2020 there was accrued R&D expense of $53,065 related to such direct expenses under this agreement.
Service Agreement 1 also estimated approximately $2.7 million (€2,172,000) in pass-through costs, mostly fees to clinical investigators and sites, which will be billed as incurred and the total contingent upon individual site rate and enrollment rates. Based on current enrollment rates and the various active clinical sites, the Company has increased its total estimated future site costs to approximately $3.1 million. This estimate could increase or decrease based on changes to individual site enrollment rates. During the year ended December 31, 2020, the Company recognized $1,317,766 in costs associated with fees to investigators and sites.
Lease Commitments – Related Party
On October 9, 2018, the Company entered into a license and membership agreement with Jelly Works X Zero-Ten, LLC for membership in a coworking space and use of an office located at 307A Kamani Street, Honolulu, HI 96813. The Company agreed to pay a monthly fee of $1,152 for membership and use of these facilities, committing to do so for a term of one year. At the end of this period, the agreement reverted to a month-to-month rental of a dedicated desk space, without office, for a monthly fee of $393 per month. In the year ended December 31, 2019, the Company rent expense associated with this agreement was approximately $12,304.
On December 19, 2018, the Company entered into an office service agreement with the University of Virginia Foundation for the use of an office and a workstation located at 1001 Research Park Boulevard, Suite 100, Charlottesville, VA 22911. The Company agreed to pay a fee of $1,150 per month for use of these facilities. The agreement is on a month-to-month basis. For the year ended December 31, 2019, the Company rent expense associated with this agreement, including continuing month-to-month payments after the expiration of the agreement, was approximately $12,650.
On March 1, 2020, the Company entered into a sublease with Purnovate, LLC, a private company in which the Company’s CEO had a 30% equity interest, for the lease of three offices at 1180 Seminole Trail, Suite 495, Charlottesville, VA 22901. The lease has a term of two years, and the monthly rent is $1,400. During the year ended December 31, 2020, the rent expense associated with this lease was $14,000.
Consulting Agreements – Related Party
On March 24, 2019, the Company entered into a consulting agreement (the “Consulting Agreement”) with Dr. Bankole A. Johnson, who at the time of the agreement was serving as the Chairman of the Board of Directors, for his service as Chief Medical Officer of the Company. The Consulting Agreement has a term of three years, unless terminated by mutual consent or by the Company for cause. Dr. Johnson resigned as Chairman of the Board of Directors at the time of execution of the consulting agreement. Under the terms of the Consulting Agreement, Dr. Johnson’s annual fee of $375,000 per year is paid twice per month. On execution, Dr. Johnson received a signing bonus of $250,000 and option to purchase 250,000 shares of common stock. Dr. Johnson’s participation in the Grant Incentive Plan (see below) and 2017 Equity Incentive Plan continue unaffected. The total expense to the Company under this agreement was $375,000 and $676,664 in the years ended December 31, 2020 and 2019, respectively.
On July 5, 2019, the Company entered into a Master Services Agreement (the “MSA”) and attached statement of work with Psychological Education Publishing Company (“PEPCO”) to administer a behavioral therapy program during the Company’s current Phase 3 clinical trial. PEPCO is owned by a related party, Dr. Bankole Johnson, the Company’s Chief Medical Officer, and currently the largest stockholder in the Company. It is anticipated that the compensation to be paid to PEPCO for services under the MSA will total approximately $300,000, of which shares of the Company’s common stock having a value equal to twenty percent (20%) of this total can be issued to Dr. Johnson in lieu of cash payment.
On December 12, 2019, the Company entered into an Amendment (the “Amendment”) to the statement of work (“SOW”). The Company had paid PEPCO $39,064 under the SOW for services rendered as of the Amendment date, leaving as estimated balance of $274,779 to be paid under the SOW. The Amendment provided the Company with a 20% discount on the remaining fees owed for services and fixed the price of any remaining services at a total of $219,823 for all services required for the use of Brief Behavioral Compliance Enhancement Treatment (BBCET) in support of the Trial. In addition, Dr. Johnson executed a guaranty, dated December 12, 2019, of PEPCO’s performance under the MSA and SOW (the “Guaranty”), together with a pledge and security agreement, dated December 12, 2019 (the “Pledge and Security Agreement”), to secure the Guaranty with 600,000 shares of the Company’s common stock beneficially owned by him and a lock-up agreement, dated December 12, 2019 (the “Lock-Up”), pursuant to which he agreed not to transfer or dispose of, directly or indirectly, any shares of the Company’s common stock, as currently owned by him, until after January 1, 2021. On August 19, 2020, the Company entered into a Lock-Up Agreement Extension and Right of First Refusal with Dr. Johnson (the “Lock-Up Extension”), which amended the Lock-Up Agreement that had been entered into dated December 12, 2019 (the “Lock-Up”). The Lock-Up Extension extended the term of Dr. Johnson’s Lock-Up from January 1, 2021 until April 1, 2021. In connection with the Lock-Up Extension, Dr. Johnson was released from his Lock-Up restrictions with respect to 350,000 shares of the Company’s common stock. During the years ended December 31, 2020 and 2019, the Company recognized $147,120 and $39,064 in expenses associated with this vendor agreement, respectively. As of December 31, 2020, the Company had recognized $147,120 in expenses, of which $108,056 were charged against cash advanced under the terms of the Amendment, leaving a net prepaid expense asset of $111,767 associated with this vendor agreement.
Other Consulting and Vendor Agreements
The Company has entered into a number of agreements and work orders for future consulting, clinical trial support, and testing services, with terms ranging between 12 and 30 months. These agreements, in aggregate, commit the Company to approximately $0.9 million in future cash.
Contingencies
The Company is subject, from time to time, to claims by third parties under various legal disputes. The defense of such claims, or any adverse outcome relating to any such claims, could have a material adverse effect on the Company’s liquidity, financial condition and cash flows. At December 31, 2020, the Company did not have any pending legal actions.
10 — SUBSEQUENT EVENTS
On January 26, 2021, the Company completed its acquisition of 100% of the equity interests of Purnovate, Inc. (See Notes 4 and 6) for cash consideration of $700,000 (including the $350,000 initial advance), the issuance 700,000 shares of Adial common stock ($2.34 at date of closing and contingent consideration for (i) certain development milestones in an aggregate amount of up to $2,100,000 for each compound (ii) milestones in an aggregate amount of up to $20,000,000 for each compound commercialized, and (iii) royalties of 3.0% of Net Sales (as defined in the Purchase Agreement). The equity consideration has been placed into escrow to secure certain indemnification and other obligations of Purnovate and the members.
The acquisition essentially included an in place workforce comprised of four employees, ongoing R&D projects and pending patents, the laboratory and office lease at 1180 Seminole Trail, Charlottesville, Virginia, laboratory equipment and glassware, and a significant library of chemical compounds that are expected to be used in their ongoing research and development projects or sold to other companies. Determination of the final fair value and purchase price allocation has not been completed as of the financial statement filing date. The Company has retained a third-party appraiser to assist in its valuation and purchase price allocation.
The Company has not presented unaudited pro forma combined results of operations as if Purnovate was acquired as of the beginning of fiscal year 2019 because it is impracticable to do so. Purnovate had no revenue and its recorded operating losses of approximately $0 and $376,000 in 2019 and 2020, respectively would not have been material to the Company’s reported losses. However, once the valuation and purchase price allocation is completed, the impact of such may be material to the pro forma combined results of operations.
On February 8, 2021, the Compensation Committee of the Board of Directors (the “Committee”) granted the Chief Executive Officer and Chief Financial Officer an option to purchase 250,000 and 125,000 shares of the Company’s common stock, respectively. The options vest over thirty-six months period and are exercisable for a period of ten years at an exercise price of $3.11. In addition, on February 8, 2021, the Committee awarded board members cumulative options to purchase 200,000 shares of the Company’s common stock vesting over thirty-six months which are exercisable for a period of ten years at $3.11.
On February 12, 2021, the Committee awarded the Chief Executive Officer and Chief Financial Officer 2020 performance bonuses of $200,000 in the aggregate. In addition, on February 12, 2021, the Company amended the CEO’s and CFO’s employment agreements to extend the term to March 31, 2026 and modify the “target bonus”, as defined.
Between February 5, 2021 and March 12, 2021, the Company exercised its right under the equity purchase agreement with Keystone Capital, LLC to direct Keystone Capital to purchase 1,007,296 shares of common stock for a total proceeds of $2,350,000, at an average purchase price of $2.33 per share.
On February 25, 2021, previously issued warrants to purchase 712,500 shares of common stock at an exercise price of $2.00 per share were exercised for a total proceeds of $1,425,000.
On March 11, 2021, the Company entered into a Securities Purchase Agreement (the “Securities Purchase Agreements”) with each of Bespoke Growth Partners, Inc. (“Bespoke”), a company controlled by Mark Peikin, the Company’s Chief Strategy Officer, three entities controlled by James W. Newman, Jr., a member of the Company’s board of directors (“Newman”), and Keystone Capital Partners, LLC (“Keystone” collectively with Bespoke and Newman, the “Investors,” and each an “Investor”), pursuant to which: (i) Bespoke agreed to purchase an aggregate of 336,667 shares of the Company’s common stock at a purchase price of $3.00 per share for aggregate gross proceeds of $1,010,001; (ii) Newman agreed to purchase an aggregate of 30,000 shares of the Company’s common stock at a purchase price of $3.00 per share for aggregate gross proceeds of $90,000; and (iii) Keystone agreed to purchase an aggregate of 333,334 shares of the Company’s common stock at a purchase price of $3.00 per share for aggregate gross proceeds of $1,000,002.
Under the terms of the Securities Purchase Agreements: (i) Bespoke agreed to purchase 33,667 shares of the Company’s common stock on the date the Securities Purchase Agreement was executed and an additional 303,000 shares of the Company’s common stock upon the effectiveness of the Registration Statement; (ii) Newman agreed to purchase 30,000 shares of the Company’s common stock on the date the Securities Purchase Agreement was executed; and (iii) Keystone agreed to purchase 33,334 shares of the Company’s common stock on the date the Securities Purchase Agreement was executed and an additional 300,000 shares of the Company’s common stock upon the effectiveness of the Registration Statement. At the date of filing, all purchases of shares effective on the date of the agreement had been completed, with fund received and shares issued.

1,575,112 SHARES OF COMMON STOCK
PROSPECTUS
, 2021
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table sets forth all expenses to be paid by the registrant, other than any estimated underwriting discounts and commissions, in connection with the offering and sale of the shares of common stock being registered. All amounts shown are estimates except for the SEC registration fee.
| | | Amount | |
| SEC registration fee | | $ | 1,121 | |
| Printing and engraving expenses | | | 5,000 | |
| Legal fees and expenses | | | 35,000 | |
| Accounting fees and expenses | | | 10,000 | |
| Transfer agent and registrar fees and expenses | | | 3,000 | |
| Miscellaneous | | | 3,879 | |
| | | | | |
| Total | | $ | 58,000 | |
Item 14. Indemnification of Directors and Officers.
The Registrant is incorporated under the laws of the State of Delaware. Section 145 of the Delaware General Corporation Law provides that a Delaware corporation may indemnify any persons who were, are, or are threatened to be made, parties to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person is or was an officer, director, employee or agent of such corporation, or is or was serving at the request of such corporation as an officer, director, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was illegal. A Delaware corporation may indemnify any persons who were, are, or are threatened to be made, a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person is or was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’\ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit provided such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests except that no indemnification is permitted without judicial approval if the officer or director is adjudged to be liable to the corporation. Where an officer or director is successful on the merits or otherwise in the defense of any action referred to above, the corporation must indemnify him or her against the expenses (including attorneys’ fees) actually and reasonably incurred.
The Registrant’s certificate of incorporation and amended and restated bylaws, each of which will become effective immediately prior to the closing of this offering, provide for the indemnification of its directors and officers to the fullest extent permitted under the Delaware General Corporation Law.
Section 102(b)(7) of the Delaware General Corporation Law permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duties as a director, except for liability for any:
| ● | transaction from which the director derives an improper personal benefit; |
| ● | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| ● | unlawful payment of dividends or redemption of shares; or |
| ● | breach of a director’s duty of loyalty to the corporation or its stockholders. |
The Registrant’s certificate of incorporation includes such a provision. Expenses incurred by any officer or director in defending any such action, suit or proceeding in advance of its final disposition shall be paid by the Registrant upon delivery to it of an undertaking, by or on behalf of such director or officer, to repay all amounts so advanced if it shall ultimately be determined that such director or officer is not entitled to be indemnified by the Registrant.
Section 174 of the Delaware General Corporation Law provides, among other things, that a director who willfully or negligently approves of an unlawful payment of dividends or an unlawful stock purchase or redemption, may be held liable for such actions. A director who was either absent when the unlawful actions were approved or dissented at the time may avoid liability by causing his or her dissent to such actions to be entered in the books containing minutes of the meetings of the board of directors at the time such action occurred or immediately after such absent director receives notice of the unlawful acts.
As permitted by the Delaware General Corporation Law, the Registrant has entered into indemnity agreements with each of its directors and executive officers, that require the Registrant to indemnify such persons against any and all costs and expenses (including attorneys’, witness or other professional fees) actually and reasonably incurred by such persons in connection with any action, suit or proceeding (including derivative actions), whether actual or threatened, to which any such person may be made a party by reason of the fact that such person is or was a director or officer or is or was acting or serving as an officer, director, employee or agent of the Registrant or any of its affiliated enterprises. Under these agreements, the Registrant is not required to provide indemnification for certain matters, including:
| ● | indemnification beyond that permitted by the Delaware General Corporation Law; |
| ● | indemnification for any proceeding with respect to the unlawful payment of remuneration to the director or officer; |
| ● | indemnification for certain proceedings involving a final judgment that the director or officer is required to disgorge profits from the purchase or sale of the Registrant’s stock; |
| ● | indemnification for proceedings involving a final judgment that the director’s or officer’s conduct was in bad faith, knowingly fraudulent or deliberately dishonest or constituted willful misconduct or a breach of his or her duty of loyalty, but only to the extent of such specific determination; |
| ● | indemnification for proceedings or claims brought by an officer or director against us or any of the Registrant’s directors, officers, employees or agents, except for claims to establish a right of indemnification or proceedings or claims approved by the Registrant’s board of directors or required by law; |
| ● | indemnification for settlements the director or officer enters into without the Registrant’s consent; or |
| ● | indemnification in violation of any undertaking required by the Securities Act or in any registration statement filed by the Registrant. |
The indemnification agreements also set forth certain procedures that will apply in the event of a claim for indemnification thereunder.
Except as otherwise disclosed under the heading “Legal Proceedings” in the “Business” section of this registration statement, there is at present no pending litigation or proceeding involving any of the Registrant’s directors or executive officers as to which indemnification is required or permitted, and the Registrant is not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
The Registrant has an insurance policy in place that covers its officers and directors with respect to certain liabilities, including liabilities arising under the Securities Act or otherwise.
Item 15. Recent Sales of Unregistered Securities.
During the last three years, we have issued unregistered securities to the persons described below. None of these transactions involved any underwriters, underwriting discounts or commissions, or any public offering. We believe that each transaction was exempt from the registration requirements of the Securities Act by virtue of Section 4(a)(2) thereof as a transaction not involving a public offering and/or Rule 3(a)(9) of the Securities Act. The recipients both had access, through their relationship with us, to information about us.
2018
On June 3, 2018, we issued to one accredited institutional investor with which Joseph Gunnar & Co., LLC had a pre-existing relationship a senior secured note in the principal amount of $325,000 (the “June 2018 Senior Note”), which is payable on March 5, 2019 or upon an earlier event of default, including, without limitation, a change of control of us. The June 2018 Senior Note is convertible into shares of our common stock at a conversion price of $2.00 per share, subject to adjustment for certain dilutive issuances. The investor also received a warrant to purchase 300,000 shares of our common stock exercisable at $3.75 per share which will be exercisable for a term of five years. The warrant provides that in the event our next financing of $2,000,000 or more includes the issuance of more than one warrant with each share of common stock sold in such next financing, then, the number of shares of common stock issuable under the warrant will be equal to 300,000 multiplied by the number of warrants sold with the common stock in the next offering.
On July 31, 2018, upon the closing of our initial public offering, approximately $310,000 aggregate principal amount of convertible debt automatically converted into an aggregate of 700,854 units, comprised of 700,854 shares of common stock and warrants to purchase 700,854 shares of common stock. The issuance of the units was exempt from registration under the Securities Act by virtue of the exemption provided under Section 3(a)(9), as the exchange was made by us with our existing security holders exclusively and no commission or other remuneration was paid or given directly or indirectly for soliciting such exchange.
On July 31, 2018, upon the closing of our initial public offering, we also issued 388,860 shares of common stock and warrants to purchase 444,608 shares of common stock to consultants and employees. The issuance of these securities was also exempt from registration under the Securities Act by virtue of Section 4(a)(2) thereof, as a transaction not involving a public offering.
On July 31, 2018, upon the closing of our initial public offering, we also issued 442,220 shares of common stock, warrants to purchase 497,330 shares of common stock, and warrants to purchase 480,600 units, each unit consisting of a share of common stock and a warrant to purchase a share of common stock debt holders. The issuance of these securities was also exempt from registration under the Securities Act by virtue of Section 4(a)(2) thereof, as a transaction not involving a public offering.
On November 12, 2018, we exchanged warrants to purchase 480,600 units having an exercise price of $5.00 per unit (each unit consisting of a share of common stock and a warrant to purchase a share of common stock at an exercise price of $6.25 per share) for two warrants, each warrant having an exercise price of $5.00, one warrant to purchase a share of common stock and a second warrant to purchase a warrant that is exercisable for a share of common stock at an exercise price of $6.25 per share. The issuance of the warrants was exempt from registration under the Securities Act by virtue of the exemption provided under Section 3(a)(9), as the exchange was made by us with our existing security holders exclusively and no commission or other remuneration was paid or given directly or indirectly for soliciting such exchange.
On November 26, 2018, we issued 100,000 shares of common stock to a consultant in consideration of strategic management consulting services and investor relations services to be rendered to us.
On December 20, 2018, we issued 162,500 shares of our common stock following receipt on December 19, 2018 of a conversion notice from the holder of an outstanding convertible note in the principal amount of $325,000, thereupon retiring all outstanding debt instruments. The issuance of common stock was exempt from registration under the Securities Act by virtue of the exemption provided under Section 3(a)(9) thereof as the common stock was exchanged by us with our existing security holder exclusively and no commission or other remuneration was paid or given directly or indirectly for soliciting such exchange.
2019
On January 21, 2019, we exchanged a currently outstanding warrant to purchase 300,000 shares of common stock exercisable at a per share price of $3.75, with a cashless exercise feature, exercisable for a period of five years from its date of issuance for a new warrant to purchase 300,000 shares of common stock at an exercise price of $3.75, with a cashless exercise feature allowing for a maximum issuance of 125,000 shares of common stock upon a cashless exercise, exercisable until April 17, 2019. Subsequent to the exchange, the warrant holder partially exercised the warrant for a payment of $468,750, for issue of 125,000 shares of common stock. The warrant holder then exercised the remainder of the warrant via a cashless and was issued 125,000 shares of common stock, retiring the warrant. We issued the new warrant and the shares of common stock upon exercise of the new warrant in reliance on the exemption from registration provided for under Section 3(a)(9) of the Securities Act, as the issuance was made to an existing security holder, there was no additional consideration paid for the new warrant or the shares of common stock and no commission or other remuneration was paid.
On January 31, 2019, the Company issued 22,311 unregistered shares of common stock upon the full cashless exercise of a warrant to purchase 65,130 shares of common stock at an exercise price of $4.99 per share.
On February 4, 2019, we issued 1,083 shares following the exercise of 1,083 previous outstanding with an exercise price of $0.005 per share, or a total exercise price of $6.
On March 6, 2019, we issued 1,083 shares following the exercise of 1,083 previous outstanding with an exercise price of $0.005 per share, or a total exercise price of $6.
On March 29, 2019, we issued 25,000 shares of common stock to a consultant at the market price of $3.43 per share.
On April 8, 2019, we issued 812 unregistered shares of common stock upon the exercise of a warrant to purchase 812 shares of common stock at an exercise price of $0.005 per share.
On May 16, 2019, we issued 4,061 unregistered shares of common stock upon the exercise of a warrant to purchase 4,061 shares of common stock at an exercise price of $0.005 per share.
On September 11, 2019, we issued 33,661 unregistered shares of common stock upon the exercise of a warrant to purchase 33,661 shares of common stock at an exercise price of $0.005 per share.
On October 29, 2019, we issued 20,305 unregistered shares of common stock upon the exercise of a warrant to purchase 33,661 shares of common stock at an exercise price of $0.005 per share.
2020
On May 15, 2020, we issued to a consultant, in consideration for services, warrant for the purchase of 72,000 shares of our common stock, with an exercise price of $1.30 per share.
On June 11, 2020, we concluded a registered direct offering of 2,820,000 shares of common stock and in a concurrent private placement the sale of warrants to purchase 2,115,000 shares of common stock at an exercise price of $2.00 per share. The shares of common stock and accompanying warrants were sold directly to the buyers at a combined at-the-market price of $1.85 for a share and three quarters warrant. Gross proceeds of the offering, totaled $5,217,000, which after offering expenses, resulted in net proceeds of $4,657,215. We issued the 2,115,000 warrants and the shares of common stock issuable upon exercise of the warrants to the investors in a concurrent private placement pursuant to an exemption from the registration requirements of the Securities Act provided in Section 4(a)(2) of the Securities Act and/or Regulation D promulgated thereunder.
On July 31, 2020, we issued warrants to purchase 1,354 shares of our common stock at an exercise price of $0.005 per share, which were exercised for a cash payment of $8.
On September 21, 2020, we entered into a securities purchase agreement with Bespoke pursuant to which it sold to Bespoke Growth Partners, Inc. 357,143 restricted shares of our common stock for an aggregate purchase price of $500,000. Bespoke’s principal is Mark H. Peikin, our Chief Strategy Officer. These shares of common stock were issued in a transaction exempt from registration under the Securities Act in reliance on Section 4(a)(2) thereof and Rule 506 of Regulation D thereunder.
On September 29, 2020, previously registered warrants to purchase 225,000 shares at an exercise fee of $2.00 per share were exercised for a total of $450,000.
On November 18, 2020, we entered into a purchase agreement (the “Purchase Agreement”) and a registration rights agreement with Keystone Capital. Pursuant to the Purchase Agreement, we have the right to sell Keystone Capital the lesser of (i) $15,000,000 in shares of our common stock and (ii) the number of shares of common stock equal to the Exchange Cap (as defined below), subject to certain limitations and conditions set forth in the Purchase Agreement, including a closing market price of our stock of greater than $1.00 on the business day sales under the agreement are made. The purchase price of the shares of our common stock that may be sold to Keystone Capital under the Purchase Agreement will be based on the market price of our common stock at the time of sale as computed under the Purchase Agreement. specifically, the purchase price per share of the common stock that may be sold to Keystone Capital under the Purchase Agreement is such fixed purchases equal ninety percent (90%) of the arithmetic average of the closing sale prices of our common stock during the five (5) consecutive trading-day period ending on the fixed purchase date for the fixed purchase, so long as the common stock is listed on Nasdaq or any nationally recognized successor thereto (to be appropriately adjusted for any reorganization, recapitalization, non-cash dividend, stock split or other similar transaction that occurs on or after the date of this Purchase Agreement). There is no upper limit on the price per share that Keystone Capital could be obligated to pay for the common stock under the Purchase Agreement.
On December 7, 2020, we closed the acquisition (the “Acquisition”) contemplated by that Equity Purchase Agreement pursuant to which we purchased all of the outstanding membership interests of Purnovate from the members of Purnovate, such that after the Acquisition, Purnovate became our wholly owned subsidiary. Purnovate is a pre-clinical drug development company with a platform focused on developing drug candidates for non-opioid pain reduction and other diseases and disorders potentially targeted with adenosine analogs that are selective, potent, stable, and soluble. Prior to the acquisition, our CEO and a Director owned equity in Purnovate and the transaction was considered to be one with a related party.
Pursuant to the terms of the Equity Purchase Agreement, in exchange for the outstanding membership interests of Purnovate, we paid the members an aggregate of $350,000 (the “Cash Consideration”) at the closing and issued to the Members an aggregate of 700,000 shares of our restricted common stock (the “Stock Consideration”) with an approximate fair market value of $1,638,000 at time of issuance, which issuance was exempt from registration pursuant to Section 4(a)(2) of the Securities Act.
2021
On March 11, 2021, we entered into Securities Purchase Agreements (the “Securities Purchase Agreements”) with each of Bespoke Growth Partners, Inc. (“Bespoke”), a company controlled by Mark Peikin, our Chief Strategy Officer, three entities controlled by James W. Newman, Jr., a member of our Board of Directors (“Newman”), and Keystone Capital Partners, LLC (“Keystone”) (collectively, the “Investors,” and each an “Investor”), pursuant to which: (i) Bespoke has agreed to purchase an aggregate of 336,667 shares of our common stock at a purchase price of $3.00 per share for aggregate gross proceeds of $1,010,001; (ii) Newman has agreed to purchase an aggregate of 30,000 shares of our common stock at a purchase price of $3.00 per share for aggregate gross proceeds of $90,000; and (iii) Keystone has agreed to purchase an aggregate of 333,334 shares of our common stock at a purchase price of $3.00 per share for aggregate gross proceeds of $1,000,002.
Under the terms of the Securities Purchase Agreements: (i) Bespoke purchased 33,667 shares of our common stock and agreed to purchase an additional 303,000 shares of our common stock upon the effectiveness of the Registration Statement; (ii) Newman purchased 30,000 shares of our common stock; and (iii) Keystone purchased 33,334 shares of our common stock and agreed to purchase an additional 300,000 shares of our common stock upon the effectiveness of a registration statement. The shares of our common stock issued, and the shares to be issued, under the Securities Purchase Agreement were, and will be, sold pursuant to an exemption from the registration requirements under Section 4(a)(2) of the Securities Act and Rule 506 of Regulation D promulgated thereunder. These Investors are accredited investors who have purchased the securities as an investment in the private placement, which did not involve a general solicitation.
We did not pay or give, directly or indirectly, any commission or other remuneration, including underwriting discounts or commissions, in connection with any of the issuances of securities listed above. The recipients of the securities in each of these transactions represented their intentions to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof, and appropriate legends were placed upon the stock certificates issued in these transactions. All recipients had adequate access, through their employment or other relationship with us or through other access to information provided by us, to information about us. The sales of these securities were made without any general solicitation or advertising.
Item 16. Exhibits and Financial Statement Schedules.
The exhibits listed in the accompanying Exhibit Index are filed or incorporated by reference as part of this registration statement.
Item 17. Undertakings.
| (a) | The undersigned registrant hereby undertakes: |
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (i) | To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933; |
| (ii) | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) (§230.424(b) of this Chapter) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement. |
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement; |
| (2) | That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| | (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| | (4) | That, for the purpose of determining liability under the Securities Act to any purchaser: If the registrant is subject to Rule 430C (§230.430C of this chapter), each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A (§230.430A of this chapter), shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use. |
| | (5) | That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser in the initial distribution of the securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: |
| | (i) | Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424 (§230.424 of this chapter); |
| | | |
| | (ii) | Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; |
| | (iii) | The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and |
| | (iv) | Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
| (b) | Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer, or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question of whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue. |
| (c) | The undersigned Registrant hereby undertakes that: |
| (1) | For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (2) | For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this Post-Effective Amendment No. 3 to the Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized in the City of Charlottesville, State of Virginia, on the 5th day of April, 2021.
| | ADIAL PHARMACEUTICALS, INC. |
| | | |
| | By: | /s/ William B. Stilley |
| | Name: | William B. Stilley |
| | Title: | President and Chief Executive Officer |
Pursuant to the requirements of the Securities Act, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature | | Title | | Date |
| | | | | |
| /s/ William B. Stilley | | Chief Executive Officer and President | | |
| William B. Stilley | | (Principal Executive Officer) | | April 5, 2021 |
| | | | | |
| /s/ Joseph M. Truluck | | Chief Operating Officer and Chief Financial Officer | | |
| Joseph M. Truluck | | (Principal Financial and Accounting Officer) | | April 5, 2021 |
| | | | | |
| * | | | | |
| J. Kermit Anderson | | Member of the Board of Directors | | April 5, 2021 |
| | | | | |
| * | | | | |
| Robertson H. Gilliland | | Member of the Board of Directors | | April 5, 2021 |
| | | | | |
| * | | | | |
| Tony Goodman | | Member of the Board of Directors | | April 5, 2021 |
| | | | | |
| * | | | | |
| James W. Newman, Jr. | | Member of the Board of Directors | | April 5, 2021 |
| | | | | |
| * | | | | |
| Kevin Schuyler, CFA | | Member of the Board of Directors | | April 5, 2021 |
| *By: | /s/ William B. Stilley | |
| | William B. Stilley
Attorney-in-Fact | |
EXHIBIT INDEX
Exhibit
Number | | Description of Exhibit |
| 3.1 | | Articles of Organization of ADial Pharmaceuticals, L.L.C. (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.2 | | Second Amended and Restated Operating Agreement of ADial Pharmaceuticals, L.L.C., dated as of February 3, 2014 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.3 | | Certificate of Incorporation of Adial Pharmaceuticals, Inc. (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.4 | | Bylaws of Adial Pharmaceuticals, Inc. (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.5 | | Articles of Incorporation of APL Conversion Corp., a Virginia Stock Corporation (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.6 | | Bylaws of APL Conversion Corp. (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.7 | | Articles of Entity Conversion of ADial Pharmaceuticals, L.L.C. filed with the Virginia Secretary of State (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.8 | | Terms and Conditions of the Plan of Entity Conversion ADial Pharmaceuticals, L.L.C. into APL Conversion Corp. (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.9 | | Certificate of Merger of Foreign Corporation into Domestic Corporation filed with the Delaware Secretary of State (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.10 | | Articles of Merger of APL Conversion Corp. into Adial Pharmaceuticals, Inc. filed with the Virginia Secretary of State (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.11 | | Agreement and Plan of Merger and Reorganization of APL Conversion Corp., a Virginia Corporation and Adial Pharmaceuticals, Inc. a Delaware Corporation (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 3.12 | | First Amendment to the Second Amended and Restated Operating Agreement of ADial Pharmaceuticals, L.L.C., dated as of September 22, 2017 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on October 25, 2017) |
| 4.1 | | Specimen Common Stock Certificate (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on October 25, 2017) |
| 4.2 | | Form of Representative’s Warrant Agreement (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 4.3 | | Form of Warrant to Purchase Membership Units (2011 Offering) (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 4.4 | | Form of Warrant to Purchase Membership Units (2013 Offering) (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 4.5 | | Form of Common Stock Purchase Warrant by and between ADial Pharmaceuticals, L.L.C and FirstFire Global Opportunities Fund, LLC (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 4.6+ | | Option Agreement between ADial Pharmaceuticals, L.L.C and Tony Goodman, effective July 1, 2017 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 4.7+ | | Grant Incentive Plan (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on April 16, 2018) |
| 4.8+ | | Form of Adial Pharmaceuticals, Inc. 2017 Equity Incentive Plan (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 4.9+ | | Form of Stock Option Grant Notice, Option Agreement (Incentive Stock Option or Nonstatutory Stock Option) and Notice of Exercise under the 2017 Equity Incentive Plan (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 4.10 | | Form of Common Stock Purchase Warrant dated November 21, 2017 by and among Adial Pharmaceuticals, Inc. and certain investors (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on November 22, 2017) |
| 4.11 | | Form of Common Stock Purchase Warrant by and between Adial Pharmaceuticals, Inc. certain investors (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on April 16, 2018) |
| 4.12 | | Form of Common Stock Purchase Warrant by and among Adial Pharmaceuticals, Inc. and consultant (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on April 16, 2018) |
| 4.13 | | Warrant to purchase 300,000 shares of Common Stock issued June 6, 2018 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on June 11, 2018) |
| 4.14 | | Form of Warrant Agent Agreement (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on June 11, 2018) |
| 4.15 | | Form of Warrant (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on June 11, 2018) |
| 4.16 | | Form of Unit Warrant (Incorporated by reference to the Company’s Form 10-Q, File No. 001-38323 filed with the Securities and Exchange Commission on September 10, 2018) |
| 4.17 | | Form of $5.00 Warrant to purchase common stock, dated November 12, 2018 (Incorporated by reference to the Company’s Form 10-Q, File No. 001-38323 filed with the Securities and Exchange Commission on November 14, 2018) |
| 4.18 | | Form of $6.25 Warrant to purchase common stock, dated November 12, 2018 (Incorporated by reference to the Company’s Form 10-Q, File No. 001-38323 filed with the Securities and Exchange Commission on November 14, 2018) |
| 4.19 | | Description of Securities (Incorporated by reference to the Company’s Annual Report on Form 10-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 22, 2021) |
| 4.20 | | Form of Common Stock Purchase Warrant (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on June 12, 2020) |
| 5.1* | | Opinion of Gracin & Marlow, LLP |
| 10.1 | | License Agreement between the University of Virginia Patent Foundation and ADial Pharmaceuticals, L.L.C. effective January 21, 2011 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 10.2 | | Amendment #1 to License Agreement between University of Virginia Patent Foundation and ADial Pharmaceuticals, L.L.C effective October 21, 2013 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 10.3 | | Amendment #2 to License Agreement between University of Virginia Patent Foundation and ADial Pharmaceuticals, L.L.C effective May 18, 2016 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 10.4 | | Amendment #3 to License Agreement between University of Virginia Patent Foundation and ADial Pharmaceuticals, L.L.C effective March 27, 2017 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 10.5+ | | Consulting Agreement between ADial Pharmaceuticals, L.L.C and Crescendo Communications, LLC Agreed to and approved June 30, 2017 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 10.6 | | Indemnification Agreement (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 10.7 | | Amendment #4 to License Agreement between University of Virginia Patent Foundation and ADial Pharmaceuticals, L.L.C effective August 15, 2017 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on September 7, 2017) |
| 10.8 | | Amendment #5 to License Agreement between University of Virginia Patent Foundation and Adial Pharmaceuticals, Inc., dated as of December 14, 2017 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on April 16, 2018) |
| 10.9 | | Security Agreement dated June 6, 2018 (Incorporated by reference to the Company’s Registration Statement on Form S-1, File No. 333-220368, filed with the Securities and Exchange Commission on June 11, 2018) |
| 10.10 | | Form of Unit Warrant (Incorporated by reference to the Company’s Form 10-Q, File No. 001-38323 filed with the Securities and Exchange Commission on September 10, 2018) |
| 10.11 | | Form of $5.00 Warrant to purchase common stock, dated November 12, 2018 (Incorporated by reference to the Company’s Form 10-Q, File No. 001-38323 filed with the Securities and Exchange Commission on November 14, 2018) |
| 10.12 | | Form of $6.25 Warrant to purchase common stock, dated November 12, 2018 (Incorporated by reference to the Company’s Form 10-Q, File No. 001-38323 filed with the Securities and Exchange Commission on November 14, 2018) |
| 10.13 | | Amendment No. 6 to License Agreement between the Company, University of Virginia Patent Foundation d/b/a the University of Virginia Licensing and Ventures Group dated as of December 18, 2018 (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on December 19, 2018) |
| 10.14 + | | Amendment to Employment Agreement between Adial Pharmaceuticals, Inc. and William B. Stilley, III, dated as of March 11, 2019 (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 14, 2019) |
| 10.15+ | | Amendment to Employment Agreement between Adial Pharmaceuticals, Inc. and Joseph Truluck, dated as of March 11, 2019 (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 14, 2019) |
| 10.16+ | | Consulting Agreement between Adial Pharmaceuticals, Inc. and Dr. Bankole Johnson, dated March 24, 2019 (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 26, 2019) |
| 10.17 | | Master Services Agreement and related statement of work, dated July 5, 2019, by and between Adial Pharmaceuticals, Inc. and Psychological Education Publishing Company (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on July 8, 2019) |
| 10.18 | | Amendment No. 1 to the Adial Pharmaceuticals, Inc. 2017 Equity Incentive Stock Plan (Incorporated by reference to the Company’s Form S-8, File No. 001-38323 filed with the Securities and Exchange Commission on September 13, 2019) |
| 10.19+ | | Form of Stock Option Grant Notice, Option Agreement (Incentive Stock Option or Nonstatutory Stock Option) and Notice of Exercise under the 2017 Equity Incentive Plan (Incorporated by reference to the Company’s Form S-8, File No. 001-38323 filed with the Securities and Exchange Commission on September 13, 2019) |
| 10.20 | | Amendment to Statement of Work under Master Services Agreement dated December 12, 2019, by and between Adial Pharmaceuticals, Inc. and Psychological Education Publishing Company (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on December 16, 2019) |
| 10.21 | | Guaranty, dated December 12, 2019, executed by Dr. Bankole Johnson (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on December 16, 2019) |
| 10.22 | | Pledge and Security Agreement, dated December 12, 2019 (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on December 16, 2019) |
| 10.23 | | Lock-Up Agreement, dated December 12, 2019 between Adial Pharmaceuticals, Inc., Bankole A. Johnson and certain entities controlled by Bankole A. Johnson (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on December 16, 2019) |
| 10.24 | | Amendment No. 7 to License Agreement by and between the University of Virginia Patent Foundation d/b/a the University of Virginia Licensing and Ventures Group and Adial Pharmaceuticals, Inc. (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on December 31, 2019) |
| 10.25+ | | Amendment to Employment Agreement between Adial Pharmaceuticals, Inc. and Joseph Truluck, dated as of March 3, 2020 (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 6, 2020) |
| 10.26 | | Placement Agency Agreement, dated June 9, 2020, by and among Adial Pharmaceuticals, Inc. and Maxim Group LLC and Joseph Gunnar & Co., LLC (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on June 10, 2020) |
| 10.27 | | Form of Securities Purchase Agreement, dated as of June 9, 2020, by and among Adial Pharmaceuticals, Inc. and the Investors (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on June 10, 2020) |
| 10.28 | | Lock-Up Agreement Extension and Right of First Refusal dated August 19, 2020 to Lock-Up Agreement (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on August 25, 2020) |
| 10.29+ | | Amendment No. 2 to the Adial Pharmaceuticals, Inc. 2017 Equity Incentive Plan (Incorporated by reference to Appendix A to the Company’s Definitive Proxy Statement on Schedule 14A filed with the Securities and Exchange Commission on July 21, 2020) |
| 10.30 | | Common Stock Purchase Agreement, dated as of November 18, 2020, by and between Adial Pharmaceuticals, Inc. and Keystone Capital Partners, LLC (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on November 24, 2020) |
| 10.31 | | Registration Rights Agreement, dated as of November 18, 2020, by and between Adial Pharmaceuticals, Inc. and Keystone Capital Partners, LLC (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on November 24, 2020) |
| 10.32 | | Equity Purchase Agreement, dated December 7, 2020, by and among Adial Pharmaceuticals, Inc., Purnovate, LLC, the members of Purnovate, LLC and Robert D. Thompson, as member representative (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on December 10, 2020) |
| 10.33+ | | Offer Letter, dated December 14, 2020 (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on December 17, 2020) |
| 10.34 | | Amendment, dated January 25, 2021, by and among Adial Pharmaceuticals, Inc., Purnovate, Inc., a wholly owned subsidiary of Adial, PNV Conversion Corp. as successor-in interest to Purnovate, LLC, and Robert D. Thompson, as member representative, to the Equity Purchase Agreement, dated December 7, 2020. (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on February 1, 2021) |
| 10.35 | | Form of Securities Purchase Agreement (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 15, 2021) |
| 10.36 | | Form of Registration Rights Agreement (Incorporated by reference to the Company’s Form 8-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 15, 2021) |
| 10.37+ | | Amendment to Executive Employment Agreement with William B. Stilley, III, effective as of February 12, 2021 (Incorporated by reference to the Company’s Annual Report on Form 10-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 22, 2021) |
| 10.38+ | | Amendment to Executive Employment Agreement with Joseph Truluck, effective as of February 12, 2021 (Incorporated by reference to the Company’s Annual Report on Form 10-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 22, 2021) |
| 10.39+ | | Amendment to Executive Employment Agreement with William B. Stilley, III, effective as of March 17, 2021 (Incorporated by reference to the Company’s Annual Report on Form 10-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 22, 2021) |
| 21.1 | | List of Subsidiaries of Adial Pharmaceuticals, Inc. (Incorporated by reference to the Company’s Annual Report on Form 10-K, File No. 001-38323 filed with the Securities and Exchange Commission on March 22, 2021) |
| 23.1** | | Consent of Friedman LLP |
| 23.2* | | Consent of Gracin & Marlow, LLP (See Exhibit 5.1 above) |
| 24.1* | | Power of Attorney (Incorporated by reference to the Company’s registration statement on Form S-1, File No. 333- 220368, filed with the Securities and Exchange Commission on April 16, 2018) |
| + | Management contract or compensatory plan or arrangement required to be identified pursuant to Item 15(a)(3) of this report. |
II-14