As filed with the Securities and Exchange Commission on May 11, 2020.
Registration No. 333-237841
Switzerland | | | 2834 | | | Not Applicable |
(State or other jurisdiction of incorporation or organization) | | | (Primary Standard Industrial Classification Code Number) | | | (I.R.S. Employer Identification No.) |
Deanna L. Kirkpatrick Yasin Keshvargar Davis Polk & Wardwell LLP 450 Lexington Avenue New York, NY 10017 (212) 450-4000 | | | Dieter Gericke Benjamin Leisinger Homburger AG Hardstrasse 201 CH-8005 Zurich, Switzerland +41 43 222 10 00 | | | Jacques Iffland Lenz & Staehelin Route de Chêne 30 CH-1211 Geneva 6, Switzerland +41 58 450 70 00 | | | Divakar Gupta Richard C. Segal Alison A. Haggerty Cooley LLP 55 Hudson Yards New York, NY 10001 (212) 479-6000 |
Title of each class of securities to be registered | | | Amount to be registered(1) | | | Proposed maximum aggregate offering price per unit(2) | | | Proposed maximum aggregate offering price(2)(3) | | | Amount of registration fee(4)(5) |
Common shares, par value CHF 0.08 per share | | | 8,458,250 | | | $18.00 | | | 152,248,500.00 | | | $19,761.86 |
| (1) | Includes 1,103,250 common shares granted pursuant to the underwriters’ option to purchase additional common shares. |
| (2) | Estimated solely for the purpose of computing the amount of the registration fee pursuant to Rule 457(a) under the Securities Act of 1933, as amended. |
| (3) | Includes the offering price of the 1,103,250 common shares granted pursuant to the underwriters’ option to purchase additional common shares. |
| (4) | Filing fees in the amount of $12,980.00 were previously paid. |
| (5) | In accordance with Rule 457(p), $6,781.86, representing the remainder of the registration fee due in connection with this Registration Statement, has been offset by the registration fee previously paid with respect to the prior registration statement on Form F-1 (No. 333-233659) initially filed by ADC Therapeutics SA on September 6, 2019. Accordingly, no additional fee is due in connection with this filing. |
| | | PER SHARE | | | TOTAL | |
Initial public offering price | | | $ | | | $ |
Underwriting discounts and commissions(1) | | | $ | | | $ |
Proceeds, before expenses, to us | | | $ | | | $ |
| (1) | See “Underwriters” for a description of all compensation payable to the underwriters. |
MORGAN STANLEY | | | BofA SECURITIES | | | COWEN |
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | |
| • | Cytotoxic Potency. The PBD dimer warheads used in our ADCs have been shown preclinically to be approximately 100 times more potent than other warheads used in currently marketed ADCs, such as auristatin, maytansine and calicheamicin. |
| • | Activity in Tumors with Low-Expressing Targets. The high potency of our PBD-based warheads means that, compared to other warheads, fewer molecules of warhead should be needed to be internalized into the cancer cell to kill it. We believe that the potency of our PBD-based warheads may allow us to develop ADCs that target antigens with low expression levels in the tumor microenvironment, potentially increasing the range of cancers amenable to treatment with ADCs. |
| • | Durable Responses. Our PBD-based ADCs create interstrand cross-links in the target cells’ DNA. As PBD cross-links are non-distortive, they are designed to be able to evade the cells’ DNA repair mechanisms that result in limited clinical responses and relapses. We believe that this may contribute to the frequency and durability of responses in heavily pre-treated and primary refractory patients that we have observed in our clinical trials. |
| • | Bystander Effect. Since our PBD-based warheads are cell-permeable, they may be able to diffuse into adjacent cells and kill them in an antigen-independent manner. We believe that this may allow us to develop ADCs that target antigens with heterogeneous expression levels in the tumor microenvironment, potentially increasing the range of cancers amenable to treatment with ADCs. |
| • | Immunogenic Cell Death. PBD warheads have been observed to induce immunogenic cell death, whereby a cancer cell’s death expresses certain stress signals that induce the body’s anti-tumor immune response through the activation of T cells and antigen-presenting cells. This opens up the potential for combining our ADCs with other therapies, particularly with immuno-oncology therapies such as checkpoint inhibitors, that are specifically designed to activate the patient’s own immune system to combat cancer. |
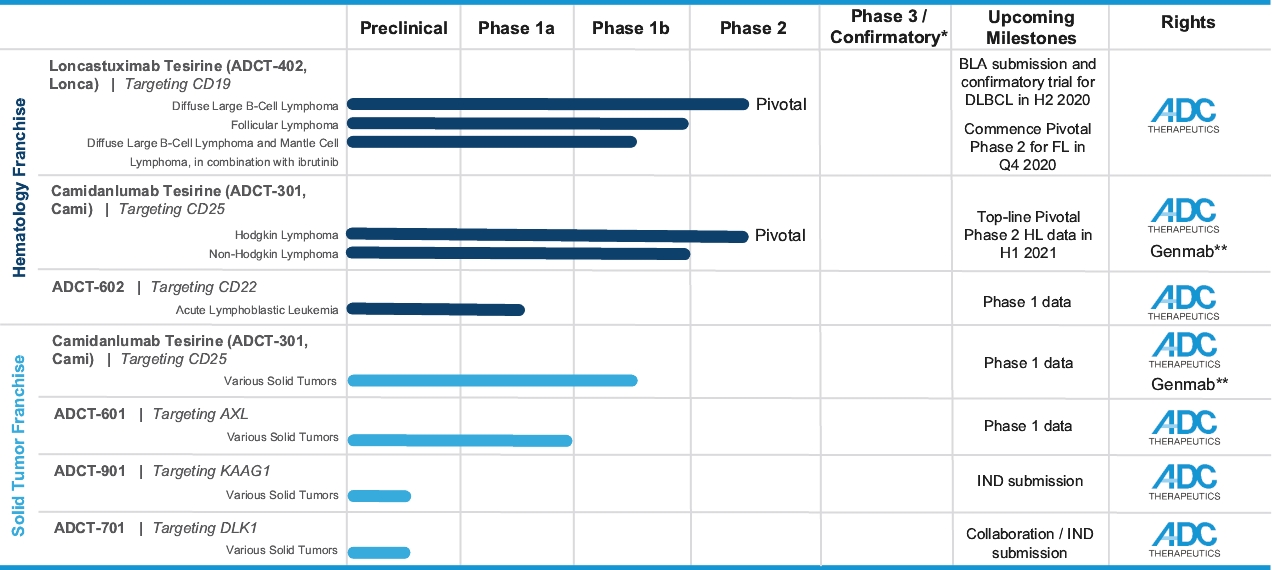
| • | We retain exclusive worldwide development and commercialization rights to Lonca. We intend to commercialize Lonca in the United States through our own infrastructure and may selectively pursue strategic collaborations in other geographies. |
| • | We intend to submit a BLA to the FDA for Lonca for the treatment of relapsed or refractory DLBCL in the second half of 2020. Concurrently, we intend to commence a post-marketing confirmatory clinical trial of Lonca in combination with rituximab, which, if successful, we believe will support a supplemental BLA for Lonca to be used as a second-line therapy for the treatment of relapsed or refractory DLBCL. |
| • | We completed enrollment of a 145-patient pivotal Phase 2 clinical trial for the treatment of relapsed or refractory DLBCL, for which we anticipate reporting final data at a medical conference in the second quarter of 2020. |
| ○ | As of October 2019, we observed a 45.5% interim ORR in 145 heavily pre-treated patients who have received a median of three prior lines of therapy. This interim ORR exceeded our target primary endpoint and the 41.4% ORR observed for DLBCL patients in our 183-patient Phase 1 clinical trial who were treated at the initial dose used in our pivotal Phase 2 clinical trial. |
| ○ | Lonca’s significant clinical activity was observed across a broad patient population in this clinical trial, including patients with primary refractory disease, bulky disease, double-hit or triple-hit disease and transformed disease, elderly patients and patients who did not respond to prior therapy. |
| • | Lonca is also being evaluated in a Phase 1/2 clinical trial in combination with ibrutinib for the treatment of relapsed or refractory DLBCL and mantle cell lymphoma (“MCL”), for which we anticipate reporting interim safety and efficacy data from the Phase 1 part of this clinical trial at a medical conference in the second quarter of 2020. |
| ○ | As of April 21, 2020, at the dose that will be used in the pivotal Phase 2 part of the clinical trial, we observed a 76.9% ORR and a 61.5% complete response rate in heavily pre-treated evaluable DLBCL patients who have received a median of three prior lines of therapy. |
| ○ | We intend to advance Lonca into a potential pivotal Phase 2 stage of this clinical trial. |
| • | We also intend to commence an additional pivotal Phase 2 clinical trial of Lonca for the treatment of FL in the fourth quarter of 2020. |
| ○ | In the Phase 1 clinical trial of Lonca for the treatment of relapsed or refractory NHL, which included 14 patients with FL, we observed a 78.6% ORR in heavily pre-treated patients with relapsed or refractory FL who have received a median of four prior lines of therapy. |
| • | Favorable clinical activity across a broad patient population, including transplant eligible and ineligible patients, patients who have not responded to first-line therapy or any prior therapy and patients with bulky disease, double-hit and triple-hit disease and transformed disease; |
| • | Significant single-agent clinical activity while maintaining a manageable tolerability profile with a low incidence of febrile neutropenia; |
| • | Activity in heavily pretreated patients, including those who have received prior CD19 therapies and stem cell transplant (“SCT”); |
| • | Promising clinical activity observed in our combination clinical trial with ibrutinib, which we believe demonstrates the opportunity to advance Lonca into earlier lines of therapy in combination with other therapies such as ibrutinib and rituximab; and |
| • | Convenient 30-minute intravenous infusions once every three weeks in the out-patient setting. |
| • | Our successful recruitment of an experienced Chief Commercial Officer and senior commercial leadership team, including a Vice President of Sales, a Vice President of Marketing and a Vice President of Market Access, with broader organizational recruitment underway; |
| • | Our successful recruitment of a Vice President of Medical Affairs, with a broader organizational recruitment underway to include field-based medical science liaisons; |
| • | Investing resources to assess the competitive landscape, supporting our differentiated profile and accelerating our launch readiness efforts; |
| • | Increased scientific interactions with Key Opinion Leaders; and |
| • | Our plans to build a U.S. field sales organization comprising of approximately 40 to 60 sales representatives, which we believe will be sufficient to reach approximately 80% of the potential prescribing physicians for Lonca. |
| • | We retain worldwide development and commercialization rights to Cami, subject to our collaboration and license agreement with Genmab. |
| • | Cami is being evaluated in a 100-patient pivotal Phase 2 clinical trial for the treatment of relapsed or refractory HL, for which we anticipate reporting top-line response rate data in the first half of 2021. |
| ○ | As of April 15, 2020, 47 patients have been enrolled in this pivotal Phase 2 clinical trial, which is currently subject to a partial clinical hold. See “Business—Camidanlumab Tesirine (ADCT-301): PBD-Based ADC Targeting CD25—Pivotal Phase 2 Clinical Trial in Relapsed or Refractory Hodgkin Lymphoma.” |
| • | We are advancing Cami through clinical development to support a BLA submission for the treatment of relapsed or refractory HL. |
| • | Enrollment has been completed in a 133-patient Phase 1 clinical trial of Cami for the treatment of relapsed or refractory HL and NHL, including 77 patients with relapsed or refractory HL. In this clinical trial, Cami demonstrated significant clinical activity across a broad patient population and maintained a tolerability profile that we believe was manageable. More specifically, as of April 2019, we observed: |
| ○ | At the initial dose for our pivotal Phase 2 clinical trial, an 86.5% ORR in heavily pre-treated patients with relapsed or refractory HL who have received a median of five prior lines of therapy, including patients who were relapsed or refractory to any or all of brentuximab vedotin, checkpoint inhibitors and stem cell transplant; and |
| ○ | A 44.0% ORR in heavily pre-treated patients with relapsed or refractory T-cell lymphoma who have received a median of four prior lines of therapy. |
| • | Cami is also being evaluated in a Phase 1b clinical trial for the treatment of selected advanced solid tumors by targeting CD25-expressing regulatory T cells (“Tregs”), for which we anticipate reporting interim data at a medical conference in the second half of 2020. |
| ○ | In paired biopsies from three patients in the Phase 1b clinical trial, we have observed a significant increase in the ratio of T effector cells (“Teffs”) to Tregs. |
| • | At the initial dose for our pivotal Phase 2 clinical trial, an 86.5% ORR in heavily pre-treated patients with relapsed or refractory HL who were relapsed or refractory to any or all of brentuximab vedotin, checkpoint inhibitors and stem cell transplant; |
| • | Tolerability profile that we believe is manageable; |
| • | The potential opportunity to advance Cami into earlier lines of therapy as a monotherapy or in combination with other therapies; |
| • | Novel immuno-oncology approach targeting Tregs for the treatment of various advanced solid tumors; and |
| • | Convenient 30-minute intravenous infusions once every three weeks in the out-patient setting. |
| • | Advance our lead product candidate, Lonca, to BLA submission in the second half of 2020. |
| • | Expand the potential market opportunity by advancing Lonca into earlier lines of therapy and for multiple indications. |
| • | Advance our second lead product candidate, Cami, to support BLA submission. |
| • | Advance our two clinical-stage solid tumor product candidates to address multiple indications in areas of high unmet medical need. |
| • | Continue to build a diverse and balanced portfolio of product candidates to address high unmet medical needs in oncology by leveraging our R&D strengths, our disciplined approach to target selection and our preclinical and clinical development strategy. |
| • | Maximize the commercial potential of our product candidates through both our own commercial organization and strategic collaborations. |
| • | We have incurred net losses during all fiscal periods since our inception, have no products approved for commercial sale and anticipate that we will continue to incur substantial net losses for the foreseeable future. |
| • | The Facility Agreement, our indebtedness and the associated restrictive covenants thereunder could adversely affect our financial condition and will restrict our ability to raise capital. |
| • | We have concentrated our research and development efforts on PBD-based ADCs, and our future success depends heavily on the successful development of this therapeutic approach. |
| • | Our current product candidates are in various stages of development, and it is possible that none of our product candidates will ever become commercial products. |
| • | Our pivotal Phase 2 clinical trial of Cami for the treatment of relapsed or refractory HL is currently subject to a partial clinical hold. In addition, in the past, certain of our clinical trials have been subject to clinical holds prior to the dosing of the first patient and partial clinical holds after the dosing of the first patient. A clinical hold on any of our clinical trials will result in delays of our clinical development timeline. |
| • | Our product candidates may cause undesirable side effects or have other properties that may delay or prevent their development or regulatory approval or limit their commercial potential. |
| • | The regulatory review and approval processes of the FDA, the European Medicines Agency (“EMA”) and comparable regulatory authorities in other jurisdictions are lengthy, time-consuming and inherently unpredictable. If we are unable to obtain, or if there are delays in obtaining, regulatory approval for our product candidates, we will not be able to commercialize our product candidates and our ability to generate revenue will be materially impaired. |
| • | As a company, we have never commercialized a product. We currently have no active sales force and we are in the process of building our commercial infrastructure. We may lack the necessary expertise, personnel and resources to successfully commercialize our product candidates. |
| • | We face substantial competition, which may result in others discovering, developing or commercializing products, treatment methods and/or technologies before or more successfully than we do. |
| • | Our rights to Cami are subject to our collaboration and license agreement with Genmab, and there can be no assurance that we will maintain the rights to develop or commercialize Cami. |
| • | We rely on third parties for the manufacture, production, storage and distribution of our product candidates. Our dependence on these third parties may impair the clinical advancement and commercialization of our product candidates. |
| • | Issued patents covering one or more of our product candidates or technologies, including Lonca, Cami or the technology we use in our product candidates, could be found invalid or unenforceable if challenged in court. |
| • | If we fail to attract and retain senior management and key scientific personnel or fail to adequately plan for succession, we may be unable to successfully develop our product candidates, conduct our clinical trials and commercialize our product candidates. |
| • | Our business could be adversely affected by the effects of health epidemics, including the recent COVID-19 pandemic, in regions where we or third parties on which we rely have significant manufacturing facilities, concentrations of clinical trial sites or other business operations. |
| • | We have identified material weaknesses in our internal control over financial reporting. If we are unable to remediate these material weaknesses or otherwise fail to maintain an effective system of internal controls, we may not be able to accurately or timely report our financial condition or results of operations, which may adversely affect our business and the price of our common shares. |
| • | a requirement to have only two years of audited financial statements in addition to any required interim financial statements and correspondingly reduced disclosure in the Management’s Discussion and Analysis of Financial Condition and Results of Operations disclosure in the registration statement of which this prospectus forms a part; |
| • | an exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”); and |
| • | to the extent that we no longer qualify as a foreign private issuer, (i) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (ii) exemptions from the requirements of holding a non-binding advisory vote on executive compensation, including golden parachute compensation. |
| • | the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; |
| • | the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and |
| • | the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K, upon the occurrence of specified significant events. |
| • | 1,020,434 common shares issuable upon the exercise of options outstanding under our 2019 Equity Incentive Plan as of December 31, 2019, at a weighted-average exercise price of $18.75 per share, of which 7,857 common shares were forfeited in the first quarter of 2020; |
| • | 1,517,339 common shares issuable upon the exercise of options outstanding under our 2019 Equity Incentive Plan granted after December 31, 2019, at a weighted-average exercise price of $18.75 per share; |
| • | Approximately 400,000 common shares issuable upon the exercise of options to be granted in connection with this offering under our 2019 Equity Incentive Plan to certain of our directors, executive officers, employees and consultants, at an exercise price per share equal to the initial public offering price in this offering, as described under “Management’s Discussion and Analysis of Financial Results and Operations—Critical Accounting Policies and Significant Judgments and Estimates—Employee Benefits”; |
| • | 7,820,000 common shares reserved for future issuance under our 2019 Equity Incentive Plan, which include the common shares issuable upon the exercise of options granted or to be granted under our 2019 Equity Incentive Plan as described in the three preceding items; |
| • | 1,538,263 common shares we hold in treasury, net of the delivery of an estimated 300,000 common shares (subject to adjustments for final tax and social security deductions) that we expect to deliver to participants of our 2014 Incentive Plan to settle outstanding awards thereunder in connection with this offering; |
| • | common shares issuable upon the exercise of options to be granted to the chairman of our board of directors in connection with this offering, as described under “Management—Compensation of Directors and Executive Officers” to bring his total rights to acquire our common shares to 2% of our then-outstanding share capital; and |
| • | common shares issuable upon the conversion of the senior secured convertible notes to be issued under the Facility Agreement. |
| • | the Share Capital Reorganization; |
| • | the filing and effectiveness of our amended and restated articles of association, which will occur immediately prior to the completion of this offering; |
| • | no exercise of the option granted to the underwriters to purchase up to 1,103,250 additional common shares in connection with this offering; |
| • | an initial public offering price of $17.00 per common share, which is the midpoint of the price range set forth on the cover page of this prospectus; |
| • | no purchase of common shares in this offering by directors, officers or existing shareholders; |
| • | the delivery to participants of our 2014 Incentive Plan of an estimated 300,000 common shares (subject to adjustments for final tax and social security deductions), which we currently hold in treasury, to settle outstanding awards under our 2014 Incentive Plan in connection with this offering; |
| • | no exercise of outstanding options after December 31, 2019, other than the settlement of outstanding awards under our 2014 Incentive Plan in connection with this offering as contemplated above; and |
| • | no conversion of the senior secured convertible notes to be issued under the Facility Agreement. |
| | | Year Ended December 31, | ||||
| | | 2019(3) | | | 2018 | |
Consolidated Income Statement Data: | | | (in USD thousands except for share and per share data) | |||
Contract revenue | | | 2,340 | | | 1,140 |
Research and development expenses | | | (107,537) | | | (118,313) |
General and administrative expenses | | | (14,202) | | | (8,768) |
Operating loss | | | (119,399) | | | (125,941) |
Other income | | | 1,655 | | | — |
Financial income | | | 2,253 | | | 2,856 |
Financial expense | | | (156) | | | — |
Exchange differences | | | (255) | | | 213 |
Loss before taxes | | | (115,902) | | | (122,872) |
Income tax expenses | | | (582) | | | (224) |
Loss for the period | | | (116,484) | | | (123,096) |
Basic and diluted loss per share(1) | | | (2.36) | | | (2.64) |
Weighted-average number of shares used to compute basic and diluted loss per share(1) | | | 49,279,961 | | | 46,600,000 |
Pro forma basic and diluted loss per share(2) | | | (2.17) | | | |
Weighted-average number of shares used to compute pro forma basic and diluted loss per share(2) | | | 53,760,234 | | | |
| (1) | See Note 28 to our audited consolidated financial statements included elsewhere in this prospectus for a description of the method used to compute basic and diluted net loss per share. These figures have been retroactively adjusted to give effect to the Share Consolidation. |
| (2) | The pro forma information gives effect to the following: (i) the delivery of 597,774 common shares by plan participants for the settlement of the 2013 Promissory Notes and the 2016 Promissory Notes; (ii) the delivery to participants of our 2014 Incentive Plan of an estimated 300,000 common shares (subject to adjustments for final tax and social security deductions), which we currently hold in treasury, to settle outstanding awards under our 2014 Incentive Plan in connection with this offering and the related payment by us of an estimated $4.0 million in tax and social security liabilities on behalf of participants of our 2014 Incentive Plan; (iii) the issuance of 4,777,996 common shares to the holders of our Class E preferred shares, as described in “Related Party Transactions—Shareholders’ Agreement”; and (iv) the Conversion, as if each such event occurred on the first day of the period presented. |
| (3) | On January 1, 2019, we adopted IFRS 16 “Leases,” the impact of which is described in Note 17 to our audited consolidated financial statements included elsewhere in this prospectus. |
| | | As of December 31, 2019 | |||||||
| | | Actual | | | Pro Forma(1) | | | Pro Forma As Adjusted(2)(3) | |
Consolidated Balance Sheet Data: | | | (in USD thousands) | ||||||
Cash and cash equivalents | | | 115,551(4) | | | 111,517 | | | 284,800 |
Total assets | | | 137,682 | | | 133,648 | | | 306,931 |
Lease liability (long-term) | | | 3,899 | | | 3,899 | | | 3,899 |
Financial liability | | | — | | | — | | | 62,000 |
Total liabilities | | | 26,526 | | | 26,526 | | | 88,526 |
Share capital | | | 4,361 | | | 4,755 | | | 5,361 |
Share premium | | | 549,922 | | | 560,678 | | | 671,354 |
Treasury shares | | | (100) | | | (11,284) | | | (11,284) |
Other reserves | | | 5,473 | | | 9,089 | | | 9,089 |
Cumulative translation adjustments | | | 69 | | | 69 | | | 69 |
Accumulated losses | | | (448,569) | | | (456,185) | | | (456,185) |
Total equity | | | 111,156 | | | 107,122 | | | 218,405 |
| (1) | The pro forma information gives effect to the following: (i) the delivery of 597,774 common shares by plan participants for the settlement of the 2013 Promissory Notes and the 2016 Promissory Notes; (ii) the delivery to participants of our 2014 Incentive Plan of an estimated 300,000 common shares (subject to adjustments for final tax and social security deductions), which we currently hold in treasury, to settle outstanding awards under our 2014 Incentive Plan in connection with this offering and the related payment by us of an estimated $4.0 million in tax and social security liabilities on behalf of participants of our 2014 Incentive Plan; (iii) the issuance of 4,777,996 common shares to the holders of our Class E preferred shares, as described in “Related Party Transactions—Shareholders’ Agreement”; and (iv) the Conversion. |
| (2) | The pro forma as adjusted information gives effect to the pro forma adjustments described in footnote (1) above and to the following: (i) our issuance and sale of 7,355,000 common shares in this offering at the assumed initial public offering price of $17.00 per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us and (ii) the receipt of the $65.0 million initial disbursement under the Facility Agreement, after deducting estimated fees and expenses payable by us, and our issuance of $65.0 million aggregate principal amount of senior secured convertible notes in connection therewith. For the purposes of the pro forma as adjusted information, the amount to be received under the Facility Agreement has been presented entirely under financial liability and has not been divided between the fair value of the embedded derivative conversion feature and the residual convertible loan, pending valuation of the embedded derivative conversion feature. The sum of the embedded derivative and the financial liability will, upon the issuance of the senior secured convertible notes, be equal to the net proceeds associated with such issuance, which, for the issuance associated with the initial disbursement under the Facility Agreement, is estimated to be approximately $62.0 million. Once the embedded derivative conversion feature has been valued, enabling the amount receivable under the Facility Agreement to be divided between the fair value of the embedded derivative conversion feature and the residual convertible loan, the $3.0 million of estimated fees and expenses payable by us will be allocated pro rata to these two components. The share of expenses allocated to the embedded derivative conversion feature will be charged directly to the consolidated income statement, while the share of expenses allocated to the residual convertible loan will be deducted from the loan. As the embedded conversion feature has not yet been valued, no such charge has been made to accumulated losses in the pro forma as adjusted figures presented above. |
| (3) | The pro forma as adjusted information is illustrative only and will change based on the actual initial public offering price and other terms of this offering determined at pricing. Each $1.00 increase in the assumed initial public offering price of $17.00 per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, would increase the pro forma as adjusted amount of each of cash and cash equivalents, total assets and total equity by $6.2 million, assuming that the number of common shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. Each $1.00 decrease in the assumed initial public offering price of $17.00 per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, would decrease the pro forma as adjusted amount of each of cash and cash equivalents, total assets and total equity by $5.9 million, assuming that the number of common shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. Each 1,000,000 share increase or decrease in the number of common shares offered by us would increase or decrease the pro forma as adjusted amount of each of cash and cash equivalents, total assets and total equity by $15.6 million, assuming the assumed initial public offering price of $17.00 per common share, the midpoint of the price range set forth on the cover page of this prospectus remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
| (4) | As of April 30, 2020, we had cash and cash equivalents of $72.5 million. |
| • | conduct and complete the pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory DLBCL; |
| • | commence and conduct a confirmatory clinical trial of Lonca in combination with rituximab for the treatment of relapsed or refractory DLBCL; |
| • | commence and conduct a pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory FL; |
| • | conduct and complete the pivotal Phase 2 clinical trial of Cami for the treatment of relapsed or refractory HL; |
| • | conduct and complete the Phase 1/2 clinical trial of Lonca and the Phase 1b clinical trial of Cami for the treatment of other indications and Phase 1 clinical trials for our other product candidates, as well as any subsequent clinical trials; |
| • | commence and conduct any required post-marketing confirmatory clinical trials for any of our product candidates in anticipation of potential accelerated approval from the FDA or similar conditional approval from the EMA or comparable regulatory agencies in other jurisdictions; |
| • | expand our research and development efforts for our preclinical product candidates and research pipeline; |
| • | seek regulatory approval for our product candidates from applicable regulatory authorities; |
| • | invest in our late-stage clinical development, manufacturing and commercialization activities, including launching commercial sales, marketing and distribution operations; |
| • | continue to prepare, file, prosecute, maintain, protect and enforce our intellectual property rights and claims; |
| • | add clinical, scientific, operational, financial and management information systems and personnel; and |
| • | operate as a public company. |
| • | the progress, results and costs of our pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory DLBCL and our pivotal Phase 2 clinical trial of Cami for the treatment of relapsed or refractory HL; |
| • | the progress, results and costs of our planned confirmatory clinical trial of Lonca in combination with rituximab for the treatment of relapsed or refractory DLBCL; |
| • | the progress, results and costs of our clinical trials of Lonca and Cami for the treatment of other indications and for our other product candidates; |
| • | the progress, results and costs of any required post-marketing confirmatory clinical trials for any product candidates that receive accelerated approval from the FDA or similar conditional approval from the EMA or comparable regulatory agencies in other jurisdictions; |
| • | the scope, progress, results and costs of researching and developing product candidates in our research pipeline, including conducting preclinical studies and clinical trials of such product candidates; |
| • | the costs of outsourced manufacturing of our product candidates, which are complex biological molecules, for clinical trials and in preparation for regulatory approval and commercialization; |
| • | the outcome, timing and costs of obtaining regulatory approvals for our product candidates if the requisite clinical trials are successful; |
| • | the size of the markets for approved indications in territories in which we receive regulatory approval, if any; |
| • | the timing and costs of commercialization activities for our product candidates, if any are approved for sale, including establishing our sales and marketing capabilities and engaging in the marketing, sales and distribution of our product candidates; |
| • | the revenue, if any, received from the commercialization of our product candidates, if any are approved for sale; |
| • | our ability to maintain and establish collaboration, licensing or other arrangements and the financial terms of such agreements; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, protecting and enforcing our intellectual property rights and claims, including any litigation costs and the outcome of such litigation; |
| • | the costs associated with potential product liability claims, including the costs associated with obtaining insurance against such claims and with defending against such claims; |
| • | the timing and amount of milestone payments we receive under our collaboration agreements; |
| • | the costs involved in maintaining and improving the technology we use in our product candidates; |
| • | our efforts to enhance operational systems and hire additional personnel, including personnel to support the development of our product candidates and to satisfy our obligations as a public company; |
| • | the effect of competing technological and market developments; and |
| • | the types of available sources of private and/or public market financing. |
| • | require us to dedicate a substantial portion of cash and cash equivalents to the payment of interest on, and principal of, the indebtedness, which will reduce the amounts available to fund working capital, capital expenditures, product development efforts and other general corporate purposes; |
| • | oblige us to negative covenants restricting our activities, including limitations on dispositions, mergers or acquisitions, encumbering our intellectual property, incurring indebtedness or liens, paying dividends, making investments and engaging in certain other business transactions; |
| • | limit our flexibility in planning for, or reacting to, changes in our business and our industry; |
| • | place us at a competitive disadvantage compared to our competitors who have less debt or competitors with comparable debt at more favorable interest rates; and |
| • | limit our ability to borrow additional amounts for working capital, capital expenditures, research and development efforts, acquisitions, debt service requirements, execution of our business strategy and other purposes and otherwise restrict our financing options. |
| • | negative preclinical data; |
| • | delays in receiving the required regulatory clearance from the appropriate regulatory authorities to commence clinical trials or amend clinical trial protocols, including any objections to our INDs or protocol amendments from the FDA; |
| • | delays in reaching, or a failure to reach, a consensus with regulatory authorities on study design; |
| • | delays in reaching, or a failure to reach, an agreement on acceptable terms with prospective clinical research organizations (“CROs”) and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and clinical trial sites; |
| • | difficulties in obtaining required Institutional Review Board (“IRB”) or ethics committee approval at each clinical trial site; |
| • | challenges in recruiting and enrolling suitable patients that meet the study criteria to participate in clinical trials; |
| • | the inability to enroll a sufficient number of patients in clinical trials to ensure adequate statistical power to detect statistically significant treatment effects; |
| • | imposition of a clinical hold by regulatory authorities or IRBs for any reason, including safety concerns and non-compliance with regulatory requirements; |
| • | failure by CROs, other third parties or us to adhere to clinical trial requirements; |
| • | failure to perform in accordance with the FDA’s good clinical practices (“GCP”) or applicable regulatory guidelines in other jurisdictions; |
| • | the inability to manufacture adequate quantities of a product candidate or other materials necessary in accordance with current Good Manufacturing Practices (“cGMPs”) to conduct clinical trials, including, for example, quality issues and delays in the testing, validation, manufacturing delays or failures at our CROs and delivery of the product candidates to the clinical trial sites; |
| • | lower than anticipated patient retention rates; |
| • | difficulties in maintaining contact with patients after treatment, resulting in incomplete data; |
| • | ambiguous or negative interim results; |
| • | our CROs or clinical trial sites failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all, deviating from the protocol or dropping out of a clinical trial; |
| • | unforeseen safety issues, including occurrence of treatment emergent adverse events (“TEAEs”) associated with the product candidate that are viewed to outweigh the product candidate’s potential benefits; |
| • | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; |
| • | lack of adequate funding to continue the clinical trial; or |
| • | delays and disruptions as a result of the COVID-19 pandemic. |
| • | the size and nature of the patient population; |
| • | the severity of the disease under investigation; |
| • | the eligibility criteria for the study in question, including any misjudgment of, and resultant adjustment to, the appropriate ranges applicable to the exclusion and inclusion criteria; |
| • | the number of clinical trial sites and the proximity of prospective patients to those sites; |
| • | the nature, severity and frequency of adverse side effects associated with our product candidates; |
| • | the standard of care in the diseases under investigation; |
| • | the commitment of our clinical investigators to identify eligible patients; |
| • | competing studies or trials with similar eligibility criteria; |
| • | the patient referral practices of physicians; |
| • | clinicians’ and patients’ perceptions as to the potential advantages and risks of the product candidate being studied in relation to other available therapies, including perception of ADCs generally and of PBD-based ADCs specifically; and |
| • | disruptions as a result of the COVID-19 pandemic. |
| • | we may encounter delays or difficulties in enrolling patients for our clinical trials due to a negative perception of our product candidates’ safety and tolerability profile; |
| • | we and/or regulatory authorities may temporarily or permanently put our clinical trials on hold; |
| • | we may be unable to obtain regulatory approval for our product candidates; |
| • | regulatory authorities may withdraw or limit their approvals of our product candidates; |
| • | regulatory authorities may require the addition of labeling statements, such as a contraindication, boxed warnings or additional warnings; |
| • | the FDA may require development of a Risk Evaluation and Mitigation Strategy with Elements to Assure Safe Use as a condition of approval; |
| • | we may decide to remove our product candidates from the marketplace; |
| • | we may be subject to regulatory investigations and government enforcement actions; |
| • | we could be sued and held liable for harm caused to patients, including as a result of hospital errors; and |
| • | our reputation may suffer. |
| • | the FDA, EMA or comparable regulatory authorities in other jurisdictions may disagree with the number, design or implementation of our clinical trials; |
| • | the population studied in the clinical trial may not be considered sufficiently broad or representative to assure safety in the full population for which we seek approval; |
| • | the FDA, EMA or comparable regulatory authorities in other jurisdictions may disagree with our interpretation of data from preclinical studies or clinical trials; |
| • | the data collected from clinical trials of our product candidates may not meet the level of statistical or clinical significance required by the FDA, EMA or comparable regulatory authorities in other jurisdictions or may otherwise not be sufficient to support the submission of a BLA, Marketing Authorization Application (“MAA”) or other submission or to obtain regulatory approval in the United States, the European Union or elsewhere; |
| • | the FDA, EMA or comparable regulatory authorities in other jurisdictions may not accept data generated by our preclinical service providers and clinical trial sites; |
| • | the FDA, EMA or comparable regulatory authorities in other jurisdictions may require us to conduct additional preclinical studies and clinical trials; |
| • | we may be unable to demonstrate to the FDA, EMA or comparable regulatory authorities in other jurisdictions that a product candidate’s response rate, DoR or risk-benefit ratio for its proposed indication is acceptable; |
| • | the FDA, EMA or comparable regulatory authorities in other jurisdictions may fail to approve the manufacturing processes, test procedures and specifications applicable to the manufacture of our product candidates, the facilities of third-party manufacturers with which we contract for clinical or commercial supplies may fail to maintain a compliance status acceptable to the FDA, EMA or comparable regulatory authorities or the EMA or comparable regulatory authorities may fail to approve facilities of third-party manufacturers with which we contract for clinical and commercial supplies; |
| • | we or any third-party service providers may be unable to demonstrate compliance with cGMPs to the satisfaction of the FDA, EMA or comparable regulatory authorities in other jurisdictions, which could result in delays in regulatory approval or require us to withdraw or recall products and interrupt commercial supply of our products; |
| • | the approval policies or regulations of the FDA, EMA or comparable regulatory authorities in other jurisdictions may change in a manner rendering our clinical data insufficient for approval; or |
| • | political factors surrounding the approval process, such as government shutdowns and political instability. |
| • | the clinical trial(s) required to verify the predicted clinical benefit of a product candidate fails to verify such benefit or does not demonstrate sufficient clinical benefit to justify the risks associated with the product candidate; |
| • | other evidence demonstrates that a product candidate is not shown to be safe or effective under the conditions of use; |
| • | we fail to conduct any required post-marketing confirmatory clinical trial with due diligence; or |
| • | we disseminate false or misleading promotional materials relating to the relevant product candidate. |
| • | restrictions on the marketing or manufacturing of the product; |
| • | withdrawal of the product from the market or voluntary or mandatory product recalls; |
| • | fines, restitution or disgorgement of profits or revenues; |
| • | warning or untitled letters; |
| • | requirements to conduct post-marketing studies or clinical trials; |
| • | holds on clinical trials; |
| • | refusal by the FDA, EMA or comparable regulatory authorities in other jurisdictions to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product license approvals; |
| • | product seizure or detention; |
| • | refusal to permit the import or export of products; and |
| • | injunctions or the imposition of civil or criminal penalties. |
| • | the safety and efficacy of the product, as demonstrated in clinical trials; |
| • | the indications for which the product is approved and the labeling approved by regulatory authorities for use with the product, including any warnings that may be required in the labeling; |
| • | our ability to offer our products for sale at competitive prices; |
| • | the perceptions of physicians, patients and patient advocacy groups of ADCs generally and PBD-based ADCs specifically; |
| • | the treatment’s cost, safety, efficacy, convenience and ease of administration compared to that of alternative treatments; |
| • | acceptance by physicians, patients and patient advocacy groups of the product as a safe and effective treatment; |
| • | the availability of coverage and adequate reimbursement by third-party payors, including cost-sharing programs such as copays and deductibles; |
| • | patients’ willingness to pay out-of-pocket in the absence of coverage and/or adequate reimbursement from third-party payors; |
| • | the effectiveness of our and our competitors’ sales and marketing efforts; |
| • | our ability to establish sales, marketing and commercial product distribution capabilities or to partner with third parties with such capabilities; |
| • | the nature, severity and frequency of adverse side effects; |
| • | any restrictions on the use of our products together with other medications; |
| • | publication of any post-approval data on the safety and effectiveness of the product; and |
| • | the success of randomized post-marketing commitment studies to confirm the benefit-risk ratio of the product. |
| • | a covered benefit under its health plan; |
| • | safe, effective and medically necessary; |
| • | appropriate for the specific patient; |
| • | cost-effective; and |
| • | neither experimental nor investigational. |
| • | increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program; |
| • | established a branded prescription drug fee that pharmaceutical manufacturers of certain branded prescription drugs must pay to the federal government; |
| • | expanded the list of covered entities eligible to participate in the 340B drug pricing program by adding new entities to the program; |
| • | established a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| • | extended manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| • | expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| • | created a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for certain drugs and biologics, including our product candidates, that are inhaled, infused, instilled, implanted or injected; |
| • | established a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; |
| • | established a Center for Medicare and Medicaid Innovation at the CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending; and |
| • | created a licensure framework for follow-on biologic products. |
| • | the demand for any products for which we may obtain regulatory approval; |
| • | our ability to set a price that we believe is fair for our products; |
| • | our ability to obtain coverage and reimbursement approval for a product; |
| • | our ability to generate revenues and achieve or maintain profitability; and |
| • | the level of taxes that we are required to pay. |
| • | advance the technology we use in our product candidates; |
| • | obtain, maintain, protect and enforce intellectual property protection for our technologies and product candidates; |
| • | obtain required government and other public and private approvals on a timely basis; |
| • | attract and retain key personnel; |
| • | execute our research and development plans; |
| • | commercialize effectively; |
| • | obtain and maintain coverage and reimbursement for our products in approved indications; |
| • | obtain adequate funding for our activities; |
| • | comply with applicable laws, regulations and regulatory requirements and restrictions with respect to the commercialization of our products, including with respect to any changed or increased regulatory restrictions; and |
| • | enter into additional strategic collaborations to advance the development and commercialization of our product candidates. |
| • | have staffing difficulties; |
| • | fail to comply with contractual obligations; |
| • | experience regulatory compliance issues; |
| • | undergo changes in priorities or become financially distressed; or |
| • | form relationships with other entities, some of which may be our competitors. |
| • | delays or stoppages in product shipments for our product candidates, including loss shipments and cross-border logistical complications, resulting in delayed and lost shipments; |
| • | delays to the development timelines for our product candidates; |
| • | an inability to commence or continue clinical trials of product candidates under development; |
| • | interruption of supply resulting from modifications to a CMO’s operations; |
| • | delays or stoppage in manufacturing or shipment due to a CMO’s bankruptcy, winding up, reorganization or similar corporate failures or financial distress; |
| • | delays in product manufacturing or shipments resulting from uncorrected defects, reliability or stability issues, or a CMO’s variation in a component; |
| • | a lack of long-term arrangements for key components; |
| • | inability to obtain adequate supply in a timely manner, or to obtain adequate supply on commercially reasonable terms; |
| • | difficulty and cost associated with locating and qualifying alternative CMOs for our components or raw materials in a timely manner; |
| • | production delays related to the evaluation and testing of components from alternative CMOs, and corresponding regulatory qualifications; |
| • | delay in delivery due to our CMOs’ prioritizing other customer orders over ours; |
| • | damage to our reputation caused by defective product candidates produced by our CMOs; and |
| • | potential price increases. |
| • | the scope of rights granted under the license agreement and other interpretation-related issues; |
| • | the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; |
| • | the sublicensing of patent and other rights under our existing collaborative development relationships and any collaboration relationships we might enter into in the future; |
| • | our diligence obligations under the license agreement and what activities satisfy those diligence obligations; |
| • | the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our current and future licensors and us; and |
| • | the priority of invention of patented technology. |
| • | others may be able to make products that are similar to any product candidates we may develop or utilize similar technology but that are not covered by the claims of the patents that we license or may own in the future; |
| • | we, or our license partners or current or future collaborators, might not have been the first to make the inventions covered by the issued patent or pending patent application that we license or may own in the future; |
| • | we, or our license partners or current or future collaborators, might not have been the first to file patent applications covering certain of our or their inventions; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our owned or licensed intellectual property rights; |
| • | it is possible that our pending licensed patent applications or those that we may own in the future will not lead to issued patents; |
| • | issued patents that we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors; |
| • | our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| • | we may not develop additional proprietary technologies that are patentable; |
| • | the patents of others may harm our business; and |
| • | we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property. |
| • | our available capital resources or capital constraints we experience; |
| • | the rate of progress, costs and results of our clinical trials and research and development activities, including the extent of scheduling conflicts with participating clinicians and collaborators; |
| • | our ability to identify and enroll patients who meet clinical trial eligibility criteria; |
| • | our receipt of approvals by the FDA, EMA and comparable regulatory authorities in other jurisdictions, and the timing thereof; |
| • | other actions, decisions or rules issued by regulators; |
| • | our ability to access sufficient, reliable and affordable supplies of materials used in the manufacture of our product candidates; |
| • | our ability to manufacture and supply clinical trial materials to our clinical sites on a timely basis; |
| • | the efforts of our collaborators with respect to the commercialization of our products; and |
| • | the securing of, costs related to, and timing issues associated with, commercial product manufacturing as well as sales and marketing activities. |
| • | The federal Anti-Kickback Statute, which prohibits any person or entity from, among other things, knowingly and willfully soliciting, receiving, offering or paying any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of an item or service reimbursable, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value. The federal Anti-Kickback Statute has also been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers, and formulary managers on the other hand. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, but the exceptions and safe harbors are drawn narrowly and require strict compliance in order to offer protection. |
| • | Federal civil and criminal false claims laws, such as the False Claims Act (“FCA”), which can be enforced by private citizens through civil qui tam actions, and civil monetary penalty laws prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented, false, fictitious or fraudulent claims for payment of federal funds, and knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to avoid, decrease or conceal an obligation to pay money to the federal government. For example, pharmaceutical companies have been prosecuted under the FCA in connection with their alleged off-label promotion of drugs, purportedly concealing price concessions in the pricing information submitted to the government for government price reporting purposes, and allegedly providing free product to customers with the expectation that the customers would bill federal healthcare programs for the product. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes “any request or demand” for money or property presented to the U.S. government. In addition, manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. |
| • | HIPAA, among other things, imposes criminal liability for executing or attempting to execute a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and creates federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services. |
| • | HIPAA, as amended by HITECH, and their implementing regulations, which impose privacy, security and breach reporting obligations with respect to individually identifiable health information upon entities subject to the law, such as health plans, healthcare clearinghouses and certain healthcare providers, known as covered entities, and their respective business associates that perform services for them that involve individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in U.S. federal courts to enforce HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. |
| • | Federal and state consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers. |
| • | The federal transparency requirements under the Physician Payments Sunshine Act, created under the Health Care Reform Act, which requires, among other things, certain manufacturers of drugs, devices, biologics and medical supplies reimbursed under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to CMS information related to payments and other transfers of value provided to physicians, as defined by such law, and teaching hospitals and physician ownership and investment interests, including such ownership and investment interests held by a physician’s immediate family members. |
| • | State and foreign laws that are analogous to each of the above federal laws, such as anti-kickback and false claims laws, that may impose similar or more prohibitive restrictions, and may apply to items or services reimbursed by non-governmental third-party payors, including private insurers. |
| • | State and foreign laws that require pharmaceutical companies to implement compliance programs, comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or to track and report gifts, compensation and other remuneration provided to physicians and other healthcare providers; state laws that require the reporting of marketing expenditures or drug pricing, including information pertaining to and justifying price increases; state and local laws that require the registration of pharmaceutical sales representatives; state laws that prohibit various marketing-related activities, such as the provision of certain kinds of gifts or meals; state laws that require the posting of information relating to clinical trials and their outcomes; and other federal, state and foreign laws that govern the privacy and security of health information or personally identifiable information in certain circumstances, including state health information privacy and data breach notification laws which govern the collection, use, disclosure, and protection of health-related and other personal information, many of which differ from each other in significant ways and often are not pre-empted by HIPAA, thus requiring additional compliance efforts. |
| • | decreased demand for our product candidates or products that we may develop; |
| • | injury to our reputation and significant negative media attention; |
| • | withdrawal of clinical trial sites and/or study participants; |
| • | significant costs to defend the related litigations; |
| • | a diversion of management’s time and our resources to pursue our business strategy; |
| • | substantial monetary awards to study participants or patients; |
| • | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| • | loss of revenue; |
| • | the inability to commercialize our product candidates that we may develop; and |
| • | a decline in the price of our common shares. |
| • | high acquisition costs; |
| • | the need to incur substantial debt or engage in dilutive issuances of equity securities to pay for acquisitions; |
| • | the potential disruption of our historical business and our activities under our collaboration agreements; |
| • | the strain on, and need to expand, our existing operational, technical, financial and administrative infrastructure; |
| • | our lack of experience in late-stage product development and commercialization; |
| • | the difficulties in assimilating employees and corporate cultures; |
| • | the difficulties in hiring qualified personnel and establishing necessary development and/or commercialization capabilities; |
| • | the failure to retain key management and other personnel; |
| • | the challenges in controlling additional costs and expenses in connection with and as a result of the acquisition; |
| • | the need to write down assets or recognize impairment charges; |
| • | the diversion of our management’s attention to integration of operations and corporate and administrative infrastructures; and |
| • | any unanticipated liabilities for activities of or related to the acquired business or its operations, products or product candidates. |
| • | economic weakness, including inflation, or political instability in particular non-U.S. economies and markets; |
| • | global trends towards pharmaceutical pricing; |
| • | differing regulatory requirements for drug approvals in non-U.S. countries; |
| • | differing reimbursement, pricing and insurance regimes; |
| • | potentially reduced protection for, and complexities and difficulties in obtaining, maintaining, protecting and enforcing, intellectual property rights; |
| • | difficulties in compliance with non-U.S. laws and regulations; |
| • | changes in non-U.S. regulations and customs, tariffs and trade barriers; |
| • | changes in non-U.S. currency exchange rates and currency controls; |
| • | changes in a specific country’s or region’s political or economic environment; |
| • | trade protection measures, economic sanctions and embargoes, import or export licensing requirements or other restrictive actions by U.S. or non-U.S. governments; |
| • | negative consequences from changes in tax laws; |
| • | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
| • | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| • | difficulties associated with staffing and managing international operations, including differing labor relations; |
| • | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; |
| • | business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters including earthquakes, typhoons, floods and fires; and |
| • | the impact of public health epidemics on employees and the global economy, such as the novel coronavirus. |
| • | results and timing of preclinical studies and clinical trials of our product candidates; |
| • | results of clinical trials of our competitors’ products; |
| • | public concern relating to the commercial value or safety of any of our product candidates; |
| • | our inability to adequately protect our proprietary rights, including patents, trademarks and trade secrets; |
| • | our inability to raise additional capital and the terms on which we raise it; |
| • | commencement or termination of any strategic collaboration or licensing arrangement; |
| • | regulatory developments, including actions with respect to our products or our competitors’ products; |
| • | actual or anticipated fluctuations in our financial condition and operating results; |
| • | publication of research reports by securities analysts about us or our competitors or our industry; |
| • | our failure or the failure of our competitors to meet analysts’ projections or guidance that we or our competitors may give to the market; |
| • | additions and departures of key personnel; |
| • | strategic decisions by us or our competitors, such as acquisitions, divestitures, spin-offs, joint ventures, strategic investments or changes in business strategy; |
| • | the passage of legislation or other regulatory developments affecting us or our industry; |
| • | fluctuations in the valuation of companies perceived by investors to be comparable to us; |
| • | sales of our common shares by us, our insiders or our other shareholders; |
| • | speculation in the press or investment community; |
| • | announcement or expectation of additional financing efforts; |
| • | any default under the Facility Agreement or the timing of any conversion of the convertible notes issued thereunder into common shares; |
| • | changes in market conditions for biopharmaceutical stocks; and |
| • | changes in general market and economic conditions. |
| • | variations in the level of expense related to the ongoing development of our product candidates or research pipeline; |
| • | results of clinical trials, or the addition or termination of clinical trials or funding support by us, or existing or future collaborators or licensing partners; |
| • | our execution of any additional collaboration, licensing or similar arrangements, and the timing of payments we may make or receive under existing or future arrangements, or the termination or modification of any such existing or future arrangements; |
| • | developments or disputes concerning patents or other proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our products; |
| • | any intellectual property infringement lawsuit or any opposition, interference, cancellation or other intellectual-property-related proceeding in which we may become involved; |
| • | additions and departures of key personnel; |
| • | strategic decisions by us or our competitors, such as acquisitions, divestitures, spin-offs, joint ventures, strategic investments or changes in business strategy; |
| • | if any of our product candidates receives regulatory approval, the terms of such approval and market acceptance and demand for such product candidates; |
| • | regulatory developments affecting our product candidates or those of our competitors; and |
| • | changes in general market and economic conditions. |
| • | the non-Swiss court had jurisdiction pursuant to the PILA; |
| • | the judgment of such non-Swiss court has become final and non-appealable; |
| • | the judgment does not contravene Swiss public policy; |
| • | the court procedures and the service of documents leading to the judgment were in accordance with the due process of law; and |
| • | no proceeding involving the same parties and the same subject matter was first brought in Switzerland, or adjudicated in Switzerland, or was earlier adjudicated in a third state, and this decision is recognizable in Switzerland. |
| • | in certain cases, allow our board of directors to place up to 32,000,000 common shares and rights to acquire an additional 32,000,000 common shares (in aggregate, approximately 50.1% of the expected outstanding share capital after completion of this offering) with affiliates or third parties, without existing shareholders having statutory pre-emptive rights in relation to this share placement; |
| • | allow our board of directors not to record any acquirer of common shares, or several acquirers acting in concert, in our share register as a shareholder with voting rights with respect to more than 15% of our share capital as set forth in the commercial register; |
| • | limit the size of our board of directors to 11 members; and |
| • | require two-thirds of the votes represented at a shareholder meeting for amending or repealing the above-mentioned voting and recording restrictions, for amending the provision setting a maximum board size or providing for indemnification of our directors and members of our executive committee and for removing the chairman or any member of the board of directors before the end of his or her term of office. |
| • | the commencement, timing, progress and results of our research and development programs, preclinical studies and clinical trials; |
| • | the timing of IND, BLA, MAA and other regulatory submissions with the FDA, EMA or comparable regulatory authorities in other jurisdictions; |
| • | the proposed clinical development pathway for our lead product candidates, Lonca and Cami, and our other product candidates, and the acceptability of the results of clinical trials for regulatory approval of such product candidates by the FDA, EMA or comparable regulatory authorities in other jurisdictions; |
| • | assumptions relating to the identification of serious adverse, undesirable or unacceptable side effects related to our product candidates; |
| • | the timing of and our ability to obtain and maintain regulatory approval for our product candidates; |
| • | our plan for the commercialization of Lonca and, subject to our collaboration and license agreement with Genmab, of Cami, if approved; |
| • | our expectations regarding the size of the patient populations amenable to treatment with our product candidates, if approved, as well as the treatment landscape of the indications that we are targeting with our product candidates; |
| • | assumptions relating to the rate and degree of market acceptance of any approved product candidates; |
| • | the pricing and reimbursement of our product candidates; |
| • | our ability to identify and develop additional product candidates; |
| • | the ability of our competitors to discover, develop or commercialize competing products before or more successfully than we do; |
| • | our competitive position and the development of and projections relating to our competitors or our industry; |
| • | our estimates of our expenses, ongoing losses, future revenue, capital requirements and our needs for or ability to obtain additional financing; |
| • | our ability to raise capital when needed in order to continue our research and development programs or commercialization efforts; |
| • | assumptions regarding the receipt of the disbursements under the Facility Agreement; |
| • | our ability to identify and successfully enter into strategic collaborations in the future, and our assumptions regarding any potential revenue that we may generate thereunder; |
| • | our ability to obtain, maintain, protect and enforce intellectual property protection for our product candidates, and the scope of such protection; |
| • | our ability to operate our business without infringing, misappropriating or otherwise violating the intellectual property rights of third parties; |
| • | our expectations regarding the impact of the COVID-19 pandemic; |
| • | our ability to attract and retain qualified key management and technical personnel; and |
| • | our expectations regarding the time during which we will be an emerging growth company under the JOBS Act and a foreign private issuer. |
| • | approximately $52 million to advance Lonca through the completion of the ongoing pivotal Phase 2 clinical trial for the treatment of relapsed or refractory DLBCL, to advance Lonca through the completion of the ongoing Phase 1/2 clinical trial in combination with ibrutinib for the treatment of relapsed or refractory DLBCL and MCL, to commence a confirmatory clinical trial of Lonca in combination with rituximab for the treatment of relapsed or refractory DLBCL, to commence a pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory FL and, if our ongoing Phase 1/2 clinical trial of Lonca in combination with ibrutinib is successful, to commence a confirmatory clinical trial of Lonca in combination with ibrutinib for the treatment of relapsed or refractory DLBCL; |
| • | approximately $14 million to advance Cami through the completion of the ongoing pivotal Phase 2 clinical trial for the treatment of relapsed or refractory HL, to advance Cami through the completion of the ongoing Phase 1b clinical trial for the treatment of selected advanced solid tumors, including the dose expansion stage in combination with a checkpoint inhibitor, and to commence potential combination clinical trials for Cami in these and other indications; |
| • | approximately $2 million to advance ADCT-602 through the completion of the ongoing Phase 1/2 clinical trial for the treatment of relapsed or refractory ALL that we are conducting with MD Anderson Cancer Center; |
| • | approximately $10 million to further advance the clinical development of ADCT-601; |
| • | approximately $100 million to further build out our commercial operations and implement our launch plans for Lonca in the United States; |
| • | approximately $19 million to fund the research and development of our preclinical product candidates and preclinical pipeline; and |
| • | the remainder for working capital and other general corporate purposes. |
| • | on an actual basis; |
| • | on a pro forma basis to give effect to the following adjustments: (i) the delivery of 597,774 common shares by plan participants for the settlement of the 2013 Promissory Notes and the 2016 Promissory Notes; (ii) the delivery to participants of our 2014 Incentive Plan of an estimated 300,000 common shares (subject to adjustments for final tax and social security deductions), which we currently hold in treasury, to settle outstanding awards under our 2014 Incentive Plan in connection with this offering and the related payment by us of an estimated $4.0 million in tax and social security liabilities on behalf of participants of our 2014 Incentive Plan; (iii) the issuance of 4,777,996 common shares to the holders of our Class E preferred shares, as described in “Related Party Transactions—Shareholders’ Agreement”; and (iv) the Conversion; and |
| • | on a pro forma as adjusted basis to give effect to the pro forma adjustments described immediately above and to the following adjustment: (i) our issuance and sale of 7,355,000 common shares in this offering at the assumed initial public offering price of $17.00 per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us; and (ii) the receipt of the $65.0 million initial disbursement under the Facility Agreement, after deducting estimated fees and expenses payable by us, and our issuance of $65.0 million aggregate principal amount of senior secured convertible notes in connection therewith. |
| | | As of December 31, 2019 | |||||||
| | | Actual | | | Pro Forma | | | Pro Forma As Adjusted(1)(2) | |
| | | (in USD thousands except for share, par value per share and per share data) | |||||||
Cash and cash equivalents | | | 115,551 | | | 111,517 | | | 284,800 |
Long-term liabilities | | | | | | | |||
Lease liability | | | 3,899 | | | 3,899 | | | 3,899 |
Financial liability | | | — | | | — | | | 62,000 |
Shareholders’ equity: | | | | | | | |||
Common shares, par value CHF 0.08 per share; 10,521,960 shares outstanding, actual; 56,577,233 shares outstanding, pro forma; 63,932,233 shares outstanding, pro forma as adjusted(3) | | | 961 | | | 4,755 | | | 5,361 |
Preferred shares, par value CHF 0.08 per share; 41,575,000 shares outstanding, actual; no shares outstanding, pro forma; no shares outstanding, pro forma as adjusted(3) | | | 3,400 | | | — | | | — |
Share premium | | | 549,922 | | | 560,678 | | | 671,354 |
Treasury shares | | | (100) | | | (11,284) | | | (11,284) |
Other reserves | | | 5,473 | | | 9,089 | | | 9,089 |
Cumulative translation adjustments | | | 69 | | | 69 | | | 69 |
Accumulated losses | | | (448,569) | | | (456,185) | | | (456,185) |
Total equity | | | 111,156 | | | 107,122 | | | 218,405 |
Total capitalization | | | 115,055 | | | 111,021 | | | 284,304 |
| (1) | For the purposes of the pro forma as adjusted information, the amount to be received under the Facility Agreement has been presented entirely under financial liability and has not been divided between the fair value of the embedded derivative conversion feature and the residual convertible loan, pending valuation of the embedded derivative conversion feature. The sum of the embedded derivative and the financial liability will, upon the issuance of the senior secured convertible notes, be equal to the net proceeds associated with such issuance, which, for the issuance associated with the initial disbursement under the Facility Agreement, is estimated to be approximately $62.0 million. Once the embedded derivative conversion feature has been valued, enabling the amount receivable under the Facility Agreement to be divided between the fair value of the embedded derivative conversion feature and the residual convertible loan, the $3.0 million of estimated fees and expenses payable by us will be allocated pro rata to these two components. The share of expenses |
| (2) | The pro forma as adjusted information is illustrative only and will change based on the actual initial public offering price and other terms of this offering determined at pricing. Each $1.00 increase in the assumed initial public offering price of $17.00 per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, would increase the pro forma as adjusted amount of each of cash and cash equivalents, total equity and total capitalization by $6.2 million, assuming that the number of common shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. Each $1.00 decrease in the assumed initial public offering price of $17.00 per common share, which is the midpoint of the price range set forth on the cover page of this prospectus, would decrease the pro forma as adjusted amount of each of cash and cash equivalents, total equity and total capitalization by $5.9 million, assuming that the number of common shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. Each 1,000,000 share increase or decrease in the number of common shares offered by us, as set forth on the cover page of this prospectus, would increase or decrease the pro forma as adjusted amount of each of cash and cash equivalents, total equity and total capitalization by $15.6 million, assuming the assumed initial public offering price of $17.00 per common share, the midpoint of the price range set forth on the cover page of this prospectus remains the same, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
| (3) | The number and par values of common shares and of preferred shares have been retroactively adjusted to give effect to the Share Consolidation. |
Assumed initial public offering price per common share | | | | | $17.00 | |
Historical net tangible book value per share as of December 31, 2019 | | | $1.97 | | | |
Decrease in net tangible book value per share as of December 31, 2019 attributable to pro forma adjustments | | | (0.23) | | | |
Pro forma net tangible book value per common share as of December 31, 2019 | | | 1.74 | | | |
Increase in pro forma net tangible book value per share attributable to new investors participating in this offering | | | 1.54 | | | |
Pro forma as adjusted net tangible book value per common share after giving effect to this offering | | | | | 3.28 | |
Dilution per common share to investors participating in this offering | | | | | $13.72 |
| | | Shares Purchased | | | Total Consideration | | | Weighted-Average Price Per Common Share | |||||||
| | | Number | | | Percent | | | Amount | | | Percent | | |||
| | | (in USD thousands except for share and per share data) | |||||||||||||
Existing shareholders before this offering | | | 56,577,233 | | | 88.5% | | | $554.1 | | | 81.6% | | | $9.79 |
Investors participating in this offering | | | 7,355,000 | | | 11.5% | | | $125.0 | | | 18.4% | | | $17.00 |
Total | | | 63,932,233 | | | 100% | | | $679.1 | | | 100% | | | |
| | | Year Ended December 31, | ||||
| | | 2019(3) | | | 2018 | |
Consolidated Income Statement Data: | | | (in USD thousands except for share and per share data) | |||
Contract revenue | | | 2,340 | | | 1,140 |
Research and development expenses | | | (107,537) | | | (118,313) |
General and administrative expenses | | | (14,202) | | | (8,768) |
Operating loss | | | (119,399) | | | (125,941) |
Other income | | | 1,655 | | | — |
Financial income | | | 2,253 | | | 2,856 |
Financial expense | | | (156) | | | — |
Exchange differences | | | (255) | | | 213 |
Loss before taxes | | | (115,902) | | | (122,872) |
Income tax expenses | | | (582) | | | (224) |
Loss for the period | | | (116,484) | | | (123,096) |
Basic and diluted loss per share(1) | | | (2.36) | | | (2.64) |
Weighted-average number of shares used to compute basic and diluted loss per share(1) | | | 49,279,961 | | | 46,600,000 |
Pro forma basic and diluted loss per share(2) | | | (2.17) | | | |
Weighted-average number of shares used to compute pro forma basic and diluted loss per share(2) | | | 53,760,234 | | | |
| (1) | See Note 28 to our audited consolidated financial statements included elsewhere in this prospectus for a description of the method used to compute basic and diluted net loss per share. These figures have been retroactively adjusted to give effect to the Share Consolidation. |
| (2) | The pro forma information gives effect to the following: (i) the delivery of 597,774 common shares by plan participants for the settlement of the 2013 Promissory Notes and the 2016 Promissory Notes; (ii) the delivery to participants of our 2014 Incentive Plan of an estimated 300,000 common shares (subject to adjustments for final tax and social security deductions), which we currently hold in treasury, to settle outstanding awards under our 2014 Incentive Plan in connection with this offering and the related payment by us of an estimated $4.0 million in tax and social security liabilities on behalf of participants of our 2014 Incentive Plan; (iii) the issuance of 4,777,996 common shares to the holders of our Class E preferred shares, as described in “Related Party Transactions—Shareholders’ Agreement”; and (iv) the Conversion, as if each such event occurred on the first day of the period presented. |
| (3) | On January 1, 2019, we adopted IFRS 16 “Leases,” the impact of which is described in Note 17 to our audited consolidated financial statements included elsewhere in this prospectus. |
| | | As of December 31, | ||||
| | | 2019 | | | 2018 | |
Consolidated Balance Sheet Data: | | | (in USD thousands) | |||
Cash and cash equivalents | | | 115,551 | | | 138,807 |
Total assets | | | 137,682 | | | 150,558 |
Share capital | | | 4,361 | | | 401 |
Share premium | | | 549,922 | | | 452,268 |
Treasury shares | | | (100) | | | — |
Other reserves | | | 5,473 | | | 5,702 |
Cumulative translation adjustments | | | 69 | | | (43) |
Accumulated losses | | | (448,569) | | | (332,085) |
Total equity | | | 111,156 | | | 126,243 |
| • | conduct and complete the pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory DLBCL; |
| • | commence and conduct a confirmatory clinical trial of Lonca in combination with rituximab for the treatment of relapsed or refractory DLBCL; |
| • | commence and conduct a pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory FL; |
| • | conduct and complete the pivotal Phase 2 clinical trial of Cami for the treatment of relapsed or refractory HL; |
| • | conduct and complete the Phase 1/2 clinical trial of Lonca and the Phase 1b clinical trial of Cami for the treatment of other indications and Phase 1 clinical trials for our other product candidates, as well as any subsequent clinical trials; |
| • | commence and conduct any required post-marketing confirmatory clinical trials for any of our product candidates in anticipation of potential accelerated approval from the FDA or similar conditional approval from the EMA or comparable regulatory agencies in other jurisdictions; |
| • | expand our research and development efforts for our preclinical product candidates and research pipeline; |
| • | seek regulatory approval for our product candidates from applicable regulatory authorities; |
| • | invest in our late-stage clinical development, manufacturing and commercialization activities, including launching commercial sales, marketing and distribution operations; |
| • | continue to prepare, file, prosecute, maintain, protect and enforce our intellectual property rights and claims; |
| • | add clinical, scientific, operational, financial and management information systems and personnel; and |
| • | operate as a public company. |
| • | salaries for research and development staff and related expenses, including share-based compensation expense; |
| • | costs for production of preclinical and clinical-stage product candidates by CMOs; |
| • | fees and other costs paid to contract research organizations in connection with the performance of preclinical studies and clinical trials; |
| • | costs of related facilities, materials and equipment; |
| • | costs associated with depreciation of right-of-use assets; |
| • | costs associated with obtaining and maintaining patents and other intellectual property; and |
| • | amortization and depreciation of tangible and intangible fixed assets used to develop our product candidates. |
| • | Lonca. Our pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory DLBCL and our Phase 1/2 clinical trial of Lonca in combination with ibrutinib for the treatment of relapsed or refractory DLBCL and MCL. |
| • | Cami. Our pivotal Phase 2 clinical trial of Cami for the treatment of relapsed or refractory HL and our Phase 1b clinical trial of Cami for the treatment of selected advanced solid tumors. |
| • | Other development programs. Our other research and development expenses related to our Phase 1/2 clinical trial of ADCT-602 for the treatment of relapsed or refractory ALL, ADCT-601 for the treatment of selected advanced solid tumors and our preclinical studies of ADCT-701 and ADCT-901 for the treatment of selected advanced solid tumors. The expenses mainly consist of salaries, costs for production of our product candidates and costs paid to contract research organizations in conjunction with clinical trials and preclinical studies. |
| | | Year Ended December 31, | | | |||||
| | | 2019 | | | 2018 | | | Change | |
| | | (in thousands) | |||||||
Lonca | | | 42,123 | | | 45,701 | | | (3,578) |
Cami | | | 31,839 | | | 29,374 | | | 2,465 |
ADCT-602 | | | 3,374 | | | 6,162 | | | (2,788) |
ADCT-601 | | | 7,183 | | | 14,250 | | | (7,067) |
MEDI3726 | | | 915 | | | 2,389 | | | (1,474) |
Preclinical product candidates and research pipeline | | | 22,103 | | | 20,437 | | | 1,666 |
Total | | | 107,537 | | | 118,313 | | | (10,776) |
| • | the scope, rate of progress, results and cost of our clinical trials, preclinical studies and other related activities; |
| • | the cost of manufacturing clinical supplies, and establishing commercial supplies, of any product candidates; |
| • | the number and characteristics of product candidates that we pursue; |
| • | the cost, timing and outcomes of regulatory approvals; |
| • | the cost and timing of establishing sales, marketing and distribution capabilities; and |
| • | the terms and timing of any collaboration, licensing or other arrangements that we may establish, including any required milestone and royalty payments thereunder. |
| | | Year Ended December 31, | | | |||||
| | | 2019 | | | 2018 | | | Change | |
| | | (in USD thousands) | |||||||
Contract revenue | | | 2,340 | | | 1,140 | | | 1,200 |
Research and development expenses | | | (107,537) | | | (118,313) | | | 10,776 |
General and administrative expenses | | | (14,202) | | | (8,768) | | | (5,434) |
Operating loss | | | (119,399) | | | (125,941) | | | 6,542 |
Other income | | | 1,655 | | | — | | | 1,655 |
Financial income | | | 2,253 | | | 2,856 | | | (603) |
Financial expense | | | (156) | | | — | | | (156) |
Exchange differences | | | (255) | | | 213 | | | (468) |
Loss before taxes | | | (115,902) | | | (122,872) | | | 6,970 |
Income tax expenses | | | (582) | | | (224) | | | (358) |
Loss for the year | | | (116,484) | | | (123,096) | | | 6,612 |
| | | Year Ended December 31, | | | |||||
| | | 2019 | | | 2018 | | | Change | |
| | | (in USD thousands) | |||||||
External costs | | | 81,363 | | | 98,493 | | | (17,130) |
Employee expenses | | | 24,916 | | | 19,246 | | | 5,670 |
Depreciation of property, plant and equipment | | | 407 | | | 330 | | | 77 |
Depreciation of right-of-use assets | | | 837 | | | — | | | 837 |
Amortization of intangible assets | | | 14 | | | 17 | | | (3) |
Impairment of intangible assets | | | — | | | 227 | | | (227) |
Research and development expenses | | | 107,537 | | | 118,313 | | | (10,776) |
| | | Year Ended December 31, | | | |||||
| | | 2019 | | | 2018 | | | Change | |
| | | (in USD thousands) | |||||||
Employee expenses | | | 5,135 | | | 4,936 | | | 199 |
External costs | | | 8,668 | | | 3,628 | | | 5,040 |
General and administrative costs charged by related parties | | | 11 | | | 38 | | | (27) |
Depreciation of property, plant and equipment | | | 145 | | | 158 | | | (13) |
Depreciation of right-of-use assets | | | 227 | | | — | | | 227 |
Amortization of intangible assets | | | 16 | | | 8 | | | 8 |
General and administrative expenses | | | 14,202 | | | 8,768 | | | 5,434 |
| | | Year Ended December 31, | | | |||||
| | | 2019 | | | 2018 | | | Change | |
| | | (in USD thousands) | |||||||
Net cash provided by (used in): | | | | | | | |||
Operating activities | | | (121,581) | | | (121,362) | | | (219) |
Investing activities | | | (2,248) | | | (2,506) | | | 258 |
Financing activities | | | 100,512 | | | (24) | | | 100,536 |
Net change in cash and cash equivalents | | | (23,317) | | | (123,892) | | | 100,575 |
| • | the progress, results and costs of our pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory DLBCL and our pivotal Phase 2 clinical trial of Cami for the treatment of relapsed or refractory HL; |
| • | the progress, results and costs of our planned confirmatory clinical trial of Lonca in combination with rituximab for the treatment of relapsed or refractory DLBCL; |
| • | the progress, results and costs of our clinical trials of Lonca and Cami for the treatment of other indications and for our other product candidates; |
| • | the progress, results and costs of any required post-marketing confirmatory clinical trials for any product candidates that receive accelerated approval from the FDA or similar conditional approval from the EMA or comparable regulatory agencies in other jurisdictions; |
| • | the scope, progress, results and costs of researching and developing product candidates in our research pipeline, including conducting preclinical studies and clinical trials of such product candidates; |
| • | the costs of outsourced manufacturing of our product candidates, which are complex biological molecules, for clinical trials and in preparation for regulatory approval and commercialization; |
| • | the outcome, timing and costs of obtaining regulatory approvals for our product candidates if the requisite clinical trials are successful; |
| • | the size of the markets for approved indications in territories in which we receive regulatory approval, if any; |
| • | the timing and costs of commercialization activities for our product candidates, if any are approved for sale, including establishing our sales and marketing capabilities and engaging in the marketing, sales and distribution of our product candidates; |
| • | the revenue, if any, received from the commercialization of our product candidates, if any are approved for sale; |
| • | our ability to maintain and establish collaboration, licensing or other arrangements and the financial terms of such agreements; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, protecting and enforcing our intellectual property rights and claims, including any litigation costs and the outcome of such litigation; |
| • | the costs associated with potential product liability claims, including the costs associated with obtaining insurance against such claims and with defending against such claims; |
| • | the timing and amount of milestone payments we receive under our collaboration agreements; |
| • | the costs involved in maintaining and improving the technology we use in our product candidates; |
| • | our efforts to enhance operational systems and hire additional personnel, including personnel to support the development and commercialization of our product candidates and to satisfy our obligations as a public company; |
| • | the effect of competing technological and market developments; and |
| • | the types of available sources of private and/or public market financing. |
| | | Payments Due By Period(1) | |||||||||||||
| | | Total | | | Less than 1 year | | | 1-3 years | | | 3-5 years | | | More than 5 years | |
| | | (in USD thousands) | |||||||||||||
Trade accounts payable | | | 3,329 | | | 3,329 | | | — | | | — | | | — |
Lease liabilities | | | 5,370 | | | 1,246 | | | 1,859 | | | 1,204 | | | 1,061 |
Total(2) | | | 8,699 | | | 4,575 | | | 1,859 | | | 1,204 | | | 1,061 |
| (1) | The amounts of contractual obligations set forth in the table above are associated with contracts that are enforceable and legally binding and that specify all significant terms, fixed or minimum services to be used, fixed, minimum or variable price provisions, and the approximate timing of the actions under the contracts. The table does not include obligations under agreements that we can cancel without a significant penalty. |
| (2) | Excludes collaboration agreements described in the preceding paragraphs above. |
| • | including any market performance conditions; |
| • | excluding the impact of any service and non-market performance vesting conditions; and |
| • | including the impact of any non-vesting conditions. |
| | | Year Ended December 31, 2019 | |
Weighted average share price (in USD) | | | 16.31 |
Strike price (in USD) | | | 18.75 |
Expected volatility (%) | | | 176.6 |
Award life (years) | | | 5.65 |
Expected dividend yield (%) | | | — |
Risk-free interest rate (%) | | | 1.67 |
| | | Year Ended December 31, | ||||
| | | 2019 | | | 2018 | |
Weighted average share price (in USD) | | | 0.51 to 16.31 | | | 0.50 to 1.25 |
Strike price (in USD) | | | 2.50 to 2.56 | | | 2.58 to 2.63 |
Expected volatility (%) | | | 176.6 to 233.1 | | | 170.0 to 233.1 |
Award life (years) | | | 0.83 to 1.29 | | | 0.83 to 1.33 |
Expected dividend yield (%) | | | — | | | — |
Risk-free interest rate (%) | | | 1.55 to 2.60 | | | 2.32 to 2.60 |
| | | Year Ended December 31, | ||||
| | | 2019 | | | 2018 | |
Weighted average share price (in USD) | | | 0.80 to 27.50 | | | 0.79 to 1.91 |
Strike price (in USD) | | | 26.00 to 28.00 | | | 25.94 |
Expected volatility (%) | | | 201.9 to 4,612.8 | | | 146.4 to 201.9 |
Award life (years) | | | 0.08 to 1.02 | | | 1.02 to 1.75 |
Expected dividend yield (%) | | | — | | | — |
Risk-free interest rate (%) | | | 2.08 to 2.57 | | | 1.84 to 2.57 |
| • | the amount of the initial measurement of lease liability; |
| • | any lease payments made at or before the commencement date less any lease incentives received; |
| • | any initial direct costs; and |
| • | restoration costs. |
| • | The embedded derivative conversion feature will be initially measured at fair value and subsequently remeasured to fair value at each reporting date. This conversion feature will be presented in the balance sheet as a liability and classified as non-equity in accordance with IFRS 9. Changes in the fair value (gains or losses) of the conversion feature at the end of each period will be recorded in the consolidated income statement. |
| • | The convertible loan’s initial fair value will be the residual amount of the consideration received, net of attributable costs, after separating out the fair value of the embedded conversion feature. The loan will be subsequently measured at its amortized cost in accordance with IFRS 9. It will be presented in the balance sheet as a financial liability. |

| • | We are a pioneer in developing highly potent and targeted PBD-based ADCs. We believe that our team, with decades of experience in this field, is well positioned to develop and commercialize PBD-based ADCs for the benefit of patients with cancer. |
| • | Our two lead product candidates, Lonca and Cami, have consistently demonstrated robust single-agent clinical activity across a broad population of heavily pre-treated patients, while maintaining tolerability profiles that we believe are manageable, and we are advancing the clinical development of Lonca to BLA submission in the second half of 2020 and of Cami to support BLA submission. |
| • | We are advancing a broad pipeline of four clinical-stage product candidates and two preclinical product candidates addressing multiple areas of unmet medical need across hematological malignancies and solid tumors, leveraging our R&D strengths, our disciplined approach to target selection and our preclinical and clinical development strategy. |
| • | We retain exclusive worldwide development and commercialization rights to all of our product candidates, other than Cami for which we have a collaboration and license agreement with Genmab. Our commercial organization has initiated pre-launch market activities and is leveraging our team’s deep industry experience to maximize the commercial potential of any approved products. |
| • | Our experienced CMC team is highly proficient in the manufacturing of PBD-based ADCs and has developed a validated commercial supply chain that has been able to consistently produce Lonca at commercial scale to support BLA submission. |
| • | Advance our lead product candidate, Lonca, to BLA submission in the second half of 2020. As of October 2019, we observed a 45.5% interim ORR in our 145-patient pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory DLBCL. This interim ORR exceeded our target primary endpoint and the 41.4% ORR observed for DLBCL patients in our 183-patient Phase 1 clinical trial who were treated at the initial dose used in our pivotal Phase 2 clinical trial. We intend to submit a BLA to the FDA in the second half of 2020. |
| • | Expand the potential market opportunity by advancing Lonca into earlier lines of therapy and for multiple indications. Based on the significant single-agent clinical activity and promising combination data with ibrutinib observed in clinical trials to date, we believe that Lonca has the opportunity to advance into earlier lines of therapy in combination with other therapies, including into first-line therapy. Concurrently with our BLA submission in the second half of 2020, we intend to commence a post-marketing confirmatory clinical trial of Lonca in combination with rituximab, which, if successful, we believe will support a supplemental BLA for Lonca to be used as a second-line therapy for the treatment of relapsed or refractory DLBCL. In addition, to further expand Lonca’s potential market opportunity, we are conducting a Phase 1/2 clinical trial of Lonca in combination with ibrutinib for the treatment of relapsed or refractory DLBCL and MCL and intend to commence a pivotal Phase 2 clinical trial of Lonca for the treatment of relapsed or refractory FL in the fourth quarter of 2020. |
| • | Advance our second lead product candidate, Cami, to support BLA submission. Based on promising results from our 133-patient Phase 1 clinical trial, we are currently evaluating Cami in a pivotal Phase 2 clinical trial for the treatment of relapsed or refractory HL that, if successful, we believe will form the basis for a BLA submission. |
| • | Advance our two clinical-stage solid tumor product candidates to address multiple indications in areas of high unmet medical need. Our PBD-based ADCs have multiple mechanisms of action that we believe make them suitable for targeting solid tumors. We are therefore evaluating Cami, which targets |
| • | Continue to build a diverse and balanced portfolio of product candidates to address high unmet medical needs in oncology by leveraging our R&D strengths, our disciplined approach to target selection and our preclinical and clinical development strategy. In addition to the lead indications that we are pursuing for Lonca and Cami, we are also evaluating these two product candidates in various other disease settings, such as MCL and FL and, in the case of Cami, multiple solid tumors, both as monotherapies and when used in combination with other therapies. Furthermore, we are advancing our other product candidates, including ADCT-601 and ADCT-602, and research programs through preclinical and clinical development. To strike an optimized risk-reward balance, we intend to address both hematological malignancies and solid tumors, across both clinically validated and novel cancer targets. |
| • | Maximize the commercial potential of our product candidates through both our own commercial organization and strategic collaborations. We currently hold exclusive worldwide development and commercialization rights to all of our product candidates, other than Cami for which we have a collaboration and license agreement with Genmab. We intend to commercialize Lonca in the United States through our own infrastructure and may selectively pursue strategic collaborations in other geographies. We have an experienced team with substantial expertise in the commercialization of oncology products to support our commercialization efforts for Lonca, if approved, which will also serve as the foundation of our U.S. commercialization efforts for our other product candidates in our hematology franchise. |

| • | Selective Targeting. Traditional chemotherapies are unable to distinguish between healthy cells and tumor cells. As a result, these therapies typically have a narrow therapeutic window (i.e., the dose range that can treat disease effectively without causing unacceptable toxic side effects). In contrast, ADCs, through their use of antigen-specific antibodies, target tumor cells or other cells in the tumor microenvironment with greater selectivity than do traditional chemotherapies. This selective targeting allows ADCs to use potent cytotoxins at dose levels that otherwise would not be tolerable. As a result, ADCs can represent a highly effective treatment approach while maintaining manageable side effects. |
| • | Wide Addressable Patient Population. ADCs represent a treatment approach that expands the treatment options available to cancer patients. Many therapies are not appropriate for certain patient populations. For example, surgery is not used when the cancer is widespread, chemotherapy may not be appropriate when the patient is too sick to tolerate or does not respond to available chemotherapeutics, stem cell transplant may not be appropriate when the patient is frail, and some novel targeted therapies such as CAR-T (i.e., a type of treatment in which a patient’s T cells are modified in the laboratory so they will attack cancer cells) may not be appropriate when there is significant comorbidity. As a result of these limitations, there remains a significant unmet medical need for patients for whom other treatment options are inappropriate or ineffective. |
| • | Potential in Relapsed or Refractory Patients. Traditional therapies typically have limited effectiveness for patients who exhibit relapsed (i.e., the cancer returns after an initial positive response to treatment) or refractory (i.e., the cancer is resistant to treatment) cancers. In contrast, some ADCs have proven efficacious in such patient populations while maintaining a manageable tolerability profile. Therefore, ADCs represent an important part of the cancer treatment paradigm, expanding the treatment options available to patients suffering from relapsed or refractory cancers. |
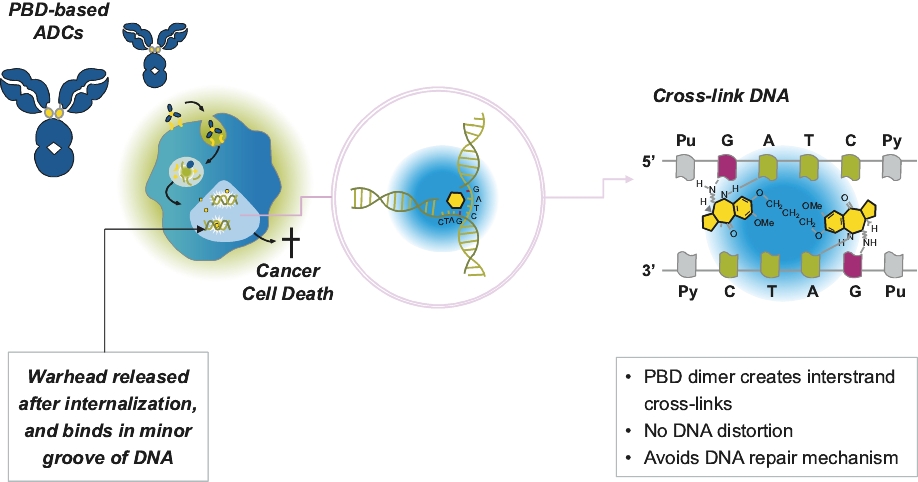
| • | Cytotoxic Potency. The PBD dimer warheads used in our ADCs have been shown preclinically to be approximately 100 times more potent than warheads used in currently marketed ADCs, such as auristatin, maytansine and calicheamicin. The figure below shows the relative in vitro cytotoxic potency of various ADC warheads and common chemotherapeutics in comparison to a PBD dimer. Despite their potency, however, the PBD dimer warheads used in our ADCs have demonstrated a manageable tolerability profile in our preclinical studies and clinical trials to date. |
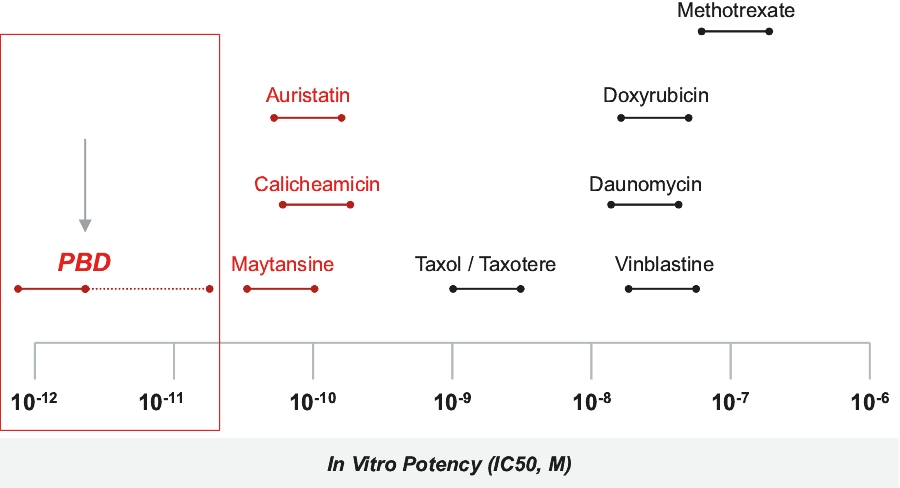
| • | Activity in Tumors with Low-Expressing Targets. Tumor cells typically require a threshold number of warhead molecules to be internalized for efficient cell killing. The high potency of our PBD-based warheads means that, compared to other warheads, fewer molecules of warhead should be needed to be internalized into the cancer cell to kill it. In cancer cells with low levels of antigen expression, ADCs with less potent warheads cannot bind in sufficient quantities to be effective. We believe that the potency of our PBD-based warheads may allow us to develop ADCs that target antigens with low expression levels in the tumor microenvironment, potentially increasing the range of cancers amenable to treatment with ADCs. |
| • | Durable Responses. Cross-links in DNA occur when an agent reacts with two nucleotides of DNA, forming a covalent linkage between them. The cross-links can occur in the same strand (i.e., intrastrand) or between opposite strands of DNA (i.e., interstrand). Our PBD-based ADCs create interstrand cross-links in the target cells’ DNA. These interstrand cross-links persist in target cells and can lie dormant, potentially for weeks. We believe that this allows our ADCs to target slowly proliferating cancer cells, including cancer stem cells. The persistence of the interstrand cross-links is explained by the fact that these cross-links do not distort the DNA helix. Cells have natural DNA repair mechanisms that detect structural changes to DNA, including those caused by cytotoxic warheads, and repair the DNA back to its original state. Warheads that create intrastrand cross-links, and even some warheads that create interstrand cross-links such as calicheamicin, distort the DNA helix, triggering the cells’ DNA repair mechanisms, thereby reducing their efficacy and leading to drug resistance. As PBD cross-links are non-distortive, they are designed to be able to evade the cells’ DNA repair mechanisms. In addition, tumor cells also induce the expression of certain transporter proteins (i.e., proteins that are able to transport warheads across the membrane outside the tumor cell) or the activation of detoxifying mechanisms that lead to inactive toxins. These potential resistance mechanisms limit traditional ADCs’ |
| • | Bystander Effect. The bystander effect occurs when a released warhead is able to diffuse into and kill neighboring cells in the tumor microenvironment, irrespective of those cells’ antigen expression. Upon binding to the target antigen and internalization of our ADCs into the tumor cell, the warhead is designed to induce apoptosis. This is followed by the release of free PBD dimers into the tumor microenvironment. Since our PBD-based warheads are cell-permeable, they may be able to diffuse into adjacent cells and kill them in an antigen-independent manner. We believe that this may allow us to develop ADCs that target antigens with heterogeneous expression levels in the tumor microenvironment, potentially increasing the range of cancers amenable to treatment with ADCs. Once the PBD is released into circulation outside the tumor microenvironment, it is rapidly excreted with a short half-life, thus limiting overall systemic toxicity. We believe that this results in our ADCs’ bystander effect being controlled and generally limited to tumor cells. |
| • | Immunogenic Cell Death. PBD warheads have been observed to induce immunogenic cell death, whereby a cancer cell’s death expresses certain stress signals that induce the body’s anti-tumor immune response through the activation of T cells and antigen-presenting cells. This opens up the potential for combining our ADCs with other therapies, particularly with immuno-oncology therapies such as checkpoint inhibitors, that are specifically designed to activate the patient’s own immune system to combat cancer. |
| • | analysis of the relative overexpression of the target antigen on the membrane of cancer cells (as compared to healthy cells) and other target characteristics, such as internalization (i.e., how rapidly the antigen migrates from the membrane to the inside of the cell), recycling (i.e., whether or not the antigen recycles back to the membrane once internalized) and shedding (i.e., whether the antigen is cleaved off from the membrane to form a soluble antigen sink in the extracellular space and/or circulation) properties; |
| • | review of whether an ADC that targets the antigen has the potential to address a clear unmet medical need and whether there is an established development path with the potential for accelerated regulatory approval; |
| • | extensive in-house research and development focused on identifying preclinical in vivo activity and on- and off-target toxicity to determine the therapeutic index of a PBD-based ADC aimed at the antigen target; and |
| • | determination of the potential product candidate’s placement in the overall risk-reward profile of our portfolio. |
| • | selecting the clinical product candidate that represents the optimal combination of antibody, linker and PBD dimer. We compare multiple candidates with different combinations of the target-specific antibodies, linkers and linker positions, conjugation chemistry and the PBD warhead. Our objective is to nominate product candidates that exhibit the optimal balance between efficacy and safety in preclinical models. |
| • | advancing our product candidates through IND-enabling preclinical studies, focusing on rapid execution of required pharmacology studies, non-clinical toxicology and pharmacokinetic studies and cGMP manufacturing of Phase 1 clinical material. Our efficient approach to preclinical development is evidenced by the following: |
| ○ | We have consistently completed IND-enabling preclinical studies in 13 to 22 months following selection of the clinical product candidate. |
| ○ | Since 2015, we have submitted five INDs and worked with our collaborators to submit two additional INDs for our product candidates. |
| • | designing clinical trials to efficiently advance our product candidates towards regulatory submission and potential approval. Our clinical trials have the following features: |
| ○ | Our Phase 1 clinical trials enroll patients with different cancers that express the target antigen on the tumor cells or other cells in the tumor microenvironment. This allows us to conduct small dose-expansion studies simultaneously with dose-escalation studies, enabling signal searching and dose selection prior to concluding Phase 1 clinical trials. We have successfully used this method in our Phase 1 clinical trials for Lonca and Cami, which resulted in the early identification of DLBCL and HL as the initial indications to pursue in our pivotal Phase 2 clinical trials, respectively. |
| ○ | Our approach allows us to identify opportunities that may expand the market opportunity for our product candidates. For example, while we are pursuing DLBCL as the lead indication for Lonca, we believe that the data generated from the Phase 1 clinical trial may allow us to more efficiently advance evaluations of Lonca for the treatment of other indications, such as MCL and FL, in pivotal clinical trials. |
| ○ | Our Phase 1 clinical trials involve a wide range of dosing regimens. Because the PBD cross-links persist in tumor cells, it is important to find the dose levels and intervals that result in optimal tumor shrinkage while minimizing cumulative toxicities due to accumulation of the cross-links between doses. The wide range of dosing regimens in our Phase 1 clinical trials enables us to select the dose level to be used in pivotal clinical trials without the need for separate dose-range finding studies. |
| ○ | Our clinical trials are designed to balance risk and reward by enrolling patients with both cancers that are difficult to treat and those that are more responsive to treatment. |
| • | encouraging close collaboration between our preclinical and clinical teams. For example, when our clinical team provides emerging pharmacokinetic data to our preclinical team, this strengthens the predictive value of our preclinical animal models when switching between indications, such as from hematological tumors to solid tumors with Cami. Our preclinical team also analyze biomarkers that correlate with patient outcomes taken by our clinical team to monitor their pharmacodynamics effects and to inform patient selection and dosing strategies. |

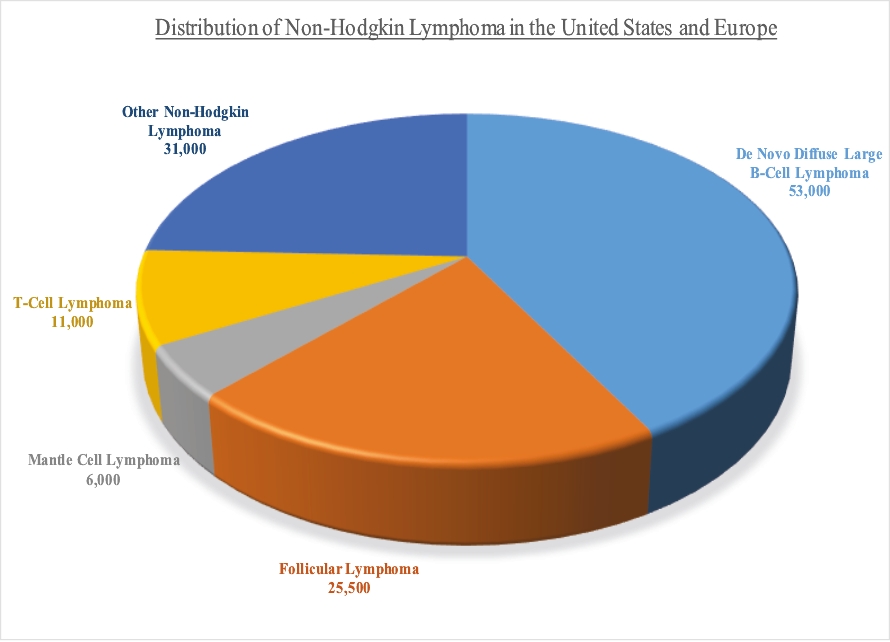
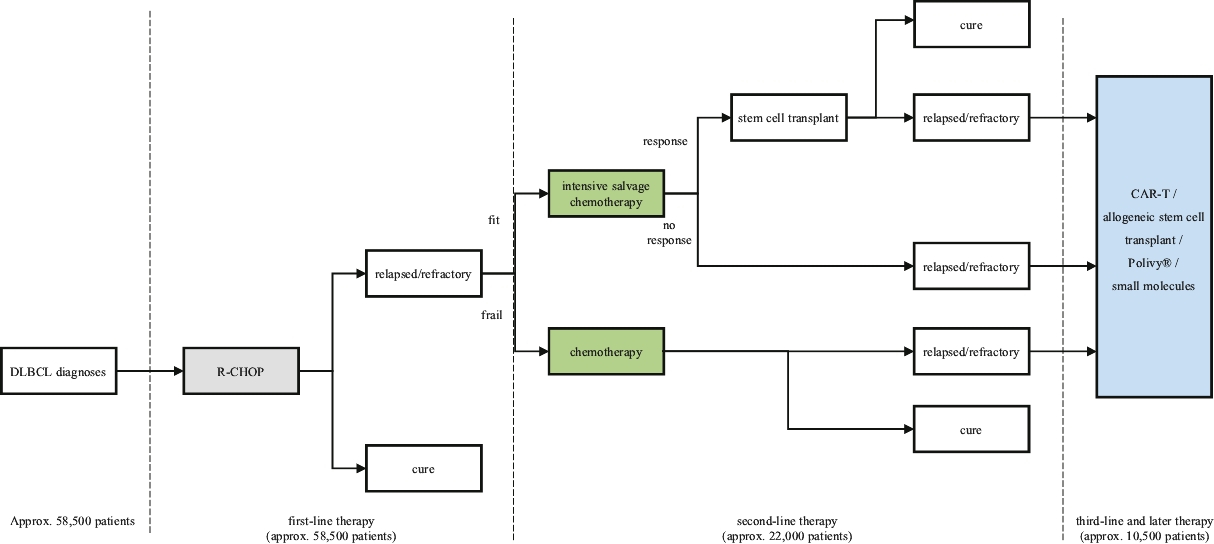
Patient Characteristics | | | Polatuzumab vedotin (Polivy®) in combination with rituximab plus bendamustine | | | Tafasitamab (MOR208) in combination with lenalidomide** |
Median Number of Prior Lines of Therapy | | | 2 | | | 2 |
SCT eligible | | | Not Included | | | Not Included |
Primary refractory disease | | | * | | | Not Included*** |
Double-hit and triple-hit DLBCL | | | Not Included | | | Not Included |
Transformed disease | | | Not Included | | | * |
Bulky disease | | | Included | | | * |
Prior treatment with CAR-T | | | * | | | Not Included |
Prior treatment with SCT | | | Included | | | Included |
Best Overall Response (%) | | | Polatuzumab vedotin (Polivy®)** | | | Tafasitamab (MOR208)** | | | Polatuzumab vedotin (Polivy®) in combination with rituximab plus bendamustine | | | Tafasitamab (MOR208) in combination with lenalidomide** |
Complete response (CR) | | | 15% | | | 6% | | | 50% | | | 43% |
Partial response (PR) | | | 37% | | | 20% | | | 13% | | | 18% |
Overall response rate (CR + PR) | | | 52% | | | 26% | | | 63% | | | 60% |
Common Grade ≥3 TEAEs (%) | | | Polatuzumab vedotin (Polivy®)** | | | Tafasitamab (MOR208)** | | | Polatuzumab vedotin (Polivy®) in combination with rituximab plus bendamustine | | | Tafasitamab (MOR208) in combination with lenalidomide** |
Neutropenia | | | 40% | | | 17% | | | 61% | | | 48% |
Anemia | | | 11% | | | 9% | | | 24% | | | * |
Peripheral sensory neuropathy | | | 9% | | | 26% | | | * | | | * |
Thrombocytopenia | | | * | | | 6% | | | 31% | | | 17% |
Lymphopenia | | | * | | | * | | | 13% | | | * |
Febrile neutropenia | | | * | | | * | | | 11% | | | 12% |
Pneumonia | | | * | | | * | | | 16% | | | 6% |
TEAEs leading to treatment discontinuation | | | * | | | * | | | 31% | | | 12% |
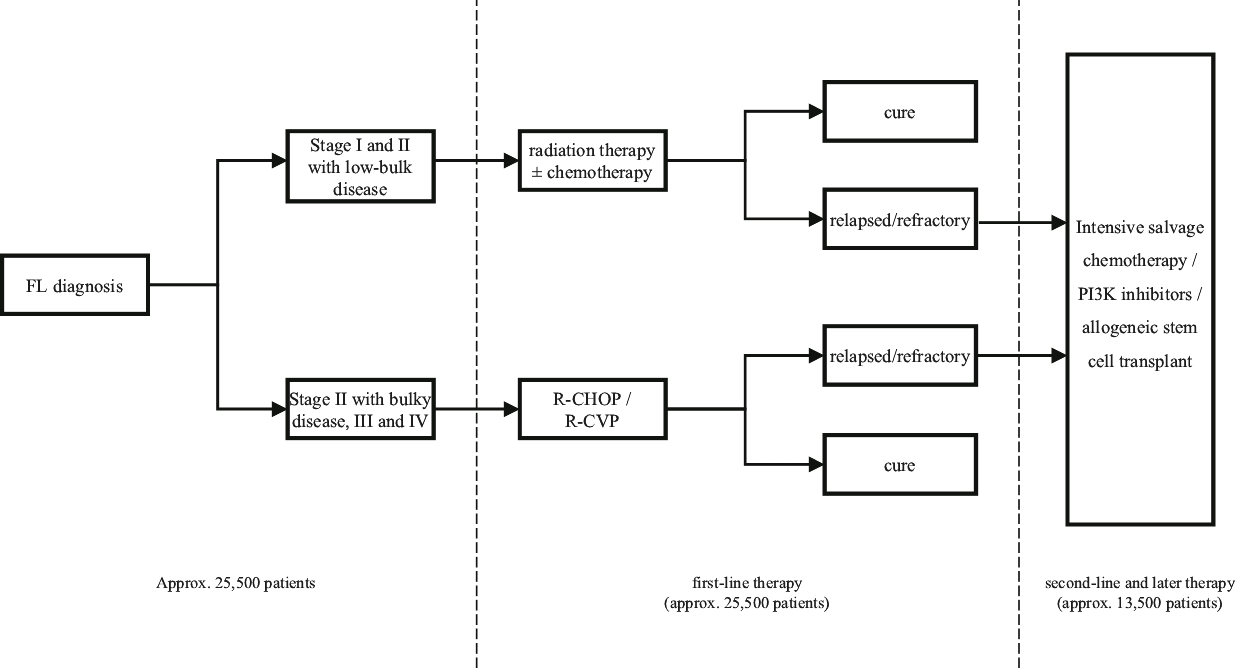
| | | Duvelisib (Copiktra®) | | | Copanlisib (Aliqopa®) | | | Idelalisib (Zydelig®) | | | Obinutuzumab (Gazyva®) in combination with bendamustine | | | Lenalidomide (Revlimid®) in combination with rituximab | |
Complete response (CR) | | | 1.2% | | | 14.4% | | | 8.3% | | | 15.5% | | | 35%-46% |
Partial response (PR) | | | 41.0% | | | 44.2% | | | 45.8% | | | 63.2% | | | 28%-46% |
Overall response rate (CR + PR) | | | 42.2% | | | 58.7% | | | 54.2% | | | 78.7% | | | 74%-80% |
Median duration of response | | | * | | | 12.2 months | | | not reached | | | not reached | | | 36.6 months - not reached |

| • | We retain exclusive worldwide development and commercialization rights to Lonca. We intend to commercialize Lonca in the United States through our own infrastructure and may selectively pursue strategic collaborations in other geographies. |
| • | We intend to submit a BLA to the FDA for Lonca for the treatment of relapsed or refractory DLBCL in the second half of 2020. Concurrently, we intend to commence a post-marketing confirmatory clinical trial of Lonca in combination with rituximab, which, if successful, we believe will support a supplemental BLA for Lonca to be used as a second-line therapy for the treatment of relapsed or refractory DLBCL. |
| • | We completed enrollment of a 145-patient pivotal Phase 2 clinical trial for the treatment of relapsed or refractory DLBCL, for which we anticipate reporting final data at a medical conference in the second quarter of 2020. |
| ○ | As of October 2019, we observed a 45.5% interim ORR in 145 heavily pre-treated patients who have received a median of three prior lines of therapy. This interim ORR exceeded our target primary endpoint and the 41.4% ORR observed for DLBCL patients in our 183-patient Phase 1 clinical trial who were treated at the initial dose used in our pivotal Phase 2 clinical trial. |
| ○ | Lonca’s significant clinical activity was observed across a broad patient population in this clinical trial, including patients with primary refractory disease, bulky disease, double-hit or triple-hit disease and transformed disease, elderly patients and patients who did not respond to prior therapy. |
| • | Lonca is also being evaluated in a Phase 1/2 clinical trial in combination with ibrutinib for the treatment of relapsed or refractory DLBCL and MCL, for which we anticipate reporting interim safety and efficacy data from the Phase 1 part of this clinical trial at a medical conference in the second quarter of 2020. |
| ○ | As of April 21, 2020, at the dose that will be used in the pivotal Phase 2 part of the clinical trial, we observed a 76.9% ORR and a 61.5% complete response rate in heavily pre-treated evaluable DLBCL patients who have received a median of three prior lines of therapy. |
| ○ | We intend to advance Lonca into a potential pivotal Phase 2 stage of this clinical trial. |
| • | We also intend to commence an additional pivotal Phase 2 clinical trial of Lonca for the treatment of FL in the fourth quarter of 2020. |
| ○ | In the Phase 1 clinical trial of Lonca for the treatment of relapsed or refractory NHL, which included 14 patients with FL, we observed a 78.6% ORR in heavily pre-treated patients with relapsed or refractory FL who have received a median of four prior lines of therapy. |
| • | Favorable clinical activity across a broad patient population, including transplant eligible and ineligible patients, patients who have not responded to first-line therapy or any prior therapy and patients with bulky disease, double-hit and triple-hit disease and transformed disease; |
| • | Significant single-agent clinical activity while maintaining a manageable tolerability profile with a low incidence of febrile neutropenia; |
| • | Activity in heavily pretreated patients, including those who have received prior CD19 therapies and SCT; |
| • | Promising clinical activity observed in our combination clinical trial with ibrutinib, which we believe demonstrates the opportunity to advance Lonca into earlier lines of therapy in combination with other therapies such as ibrutinib and rituximab; and |
| • | Convenient 30-minute intravenous infusions once every three weeks in the out-patient setting. |
| • | Our successful recruitment of an experienced Chief Commercial Officer and senior commercial leadership team, including a Vice President of Sales, a Vice President of Marketing and a Vice President of Market Access, with broader organizational recruitment underway; |
| • | Our successful recruitment of a Vice President of Medical Affairs, with a broader organizational recruitment underway to include field-based medical science liaisons; |
| • | Investing resources to assess the competitive landscape, supporting our differentiated profile and accelerating our launch readiness efforts; |
| • | Increased scientific interactions with Key Opinion Leaders; and |
| • | Our plans to build a U.S. field sales organization comprising of approximately 40 to 60 sales representatives, which we believe will be sufficient to reach approximately 80% of the potential prescribing physicians for Lonca. |
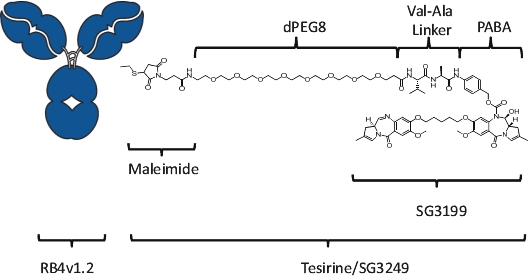
| • | CD19 is a clinically validated target for the treatment of B cell malignancies. |
| • | CD19 is expressed in B cell lineage at an earlier stage compared to CD20, which is another well-known target for the treatment of hematological malignancies. |
| • | The CD19 antigen is rapidly internalized by the cell. Therefore, it is an effective target for ADC therapy since ADCs bind only to antigens on the cell surface and the ADCs must be internalized to release the warhead inside the cell. |
| • | The CD19 antigen does not shed into the circulation. Therefore, there are no, or very low, levels of soluble CD19 to compete for binding of the ADC. |
Patient Characteristics | | | | | | | |||
Age, median (minimum, maximum) | | | 63 | | | (20, 87) | |||
Number of previous chemotherapies received, median (minimum, maximum) | | | 3 | | | (1, 13) | |||
Response to first-line chemotherapy, n (%) | | | Relapsed | | | 115 | | | (62.8) |
| | | Refractory | | | 43 | | | (23.5) | |
Response to last-line chemotherapy, n (%) | | | Relapsed | | | 66 | | | (36.1) |
| | | Refractory | | | 109 | | | (59.6) | |
Prior stem cell transplant, n (%) | | | Yes | | | 42 | | | (23.0) |
| | | No | | | 141 | | | (77.0) | |
| • | The MTD was not reached in the dose escalation stage. |
| • | Grade ≥3 TEAEs were reported in 108 patients, or 77.7% of patients. The table below presents the most common Grade ≥3 TEAEs that were reported in more than 5% of patients. |
Grade ≥3 TEAEs*, n (%) | | | Dose Levels | ||||||||||||
| | ≤90 µg/kg (n=10) | | | 120 µg/kg (n=32) | | | 150 µg/kg (n=70) | | | 200 µg/kg (n=27) | | | All Doses (n=139) | ||
Neutrophil count decreased** | | | 2 (20.0) | | | 10 (31.2) | | | 26 (37.1) | | | 15 (55.6) | | | 53 (38.1) |
Platelet count decreased** | | | 1 (10.0) | | | 7 (21.9) | | | 18 (25.7) | | | 11 (40.7) | | | 37 (26.6) |
Gamma-glutamyltransferase increased | | | 1 (10.0) | | | 7 (21.9) | | | 12 (17.1) | | | 7 (25.9) | | | 27 (19.4) |
Anemia | | | 2 (20.0) | | | 3 (9.4) | | | 11 (15.7) | | | 3 (11.1) | | | 19 (13.7) |
Disease progression | | | 0 (0.0) | | | 2 (6.3) | | | 8 (11.4) | | | 0 (0.0) | | | 10 (7.2) |
Hypokalemia | | | 0 (0.0) | | | 0 (0.0) | | | 8 (11.4) | | | 1 (3.7) | | | 9 (6.5) |
Alkaline phosphatase increased | | | 1 (10.0) | | | 3 (9.4) | | | 3 (4.3) | | | 1 (3.7) | | | 8 (5.8) |
Lymphocyte count decreased | | | 0 (0.0) | | | 2 (6.3) | | | 4 (5.7) | | | 2 (7.4) | | | 8 (5.8) |
Alanine aminotransferase increased | | | 0 (0.0) | | | 2 (6.3) | | | 3 (4.3) | | | 2 (7.4) | | | 7 (5.0) |
Fatigue | | | 0 (0.0) | | | 2 (6.3) | | | 2 (2.9) | | | 3 (11.1) | | | 7 (5.0) |
White blood cell count decreased | | | 0 (0.0) | | | 2 (6.3) | | | 3 (4.3) | | | 2 (7.4) | | | 7 (5.0) |
Patients with any Grade ≥3 TEAEs | | | 4 (40.0) | | | 24 (75.0) | | | 56 (80.0) | | | 24 (88.9) | | | 108 (77.7) |
| • | TEAEs in 26 patients, or 18.7% of patients, led to treatment discontinuation. |
| • | Across all dose levels, 32 patients, or 23.4% of patients, achieved a complete response and another 26 patients, or 19.0% of patients, achieved a partial response, resulting in a 42.3% ORR. At dose levels ≥120 μg/kg, 31 patients, or 24.4% of patients, achieved a complete response and another 25 patients, or 19.7% of patients, achieved a partial response, resulting in a 44.1% ORR. The table below shows the response rate data from this clinical trial. |
Best Overall Response, n (%) | | | Dose Levels | ||||||||||||
| | ≤90 µg/kg (n=10) | | | 120 µg/kg (n=32) | | | 150 µg/kg (n=70) | | | 200 µg/kg (n=25) | | | All Doses (n=137) | ||
Complete response (CR) | | | 1 (10.0) | | | 6 (18.8) | | | 15 (21.4) | | | 10 (40.0) | | | 32 (23.4) |
Partial response (PR) | | | 1 (10.0) | | | 8 (25.0) | | | 14 (20.0) | | | 3 (12.0) | | | 26 (19.0) |
Stable disease | | | 2 (20.0) | | | 7 (21.9) | | | 12 (17.1) | | | 2 (8.0) | | | 23 (16.8) |
Progressive disease | | | 6 (60.0) | | | 10 (31.3) | | | 28 (40.0) | | | 10 (40.0) | | | 54 (39.4) |
Not evaluable | | | 0 (0.0) | | | 1 (3.1) | | | 1 (1.4) | | | 0 (0.0) | | | 2 (1.5) |
Overall response rate (CR + PR) | | | 2 (20.0) | | | 14 (43.8) | | | 29 (41.4) | | | 13 (52.0) | | | 58 (42.3) |
| • | In our post-hoc subset analysis that excluded patients with double-hit or triple-hit disease and patients with primary refractory and early relapsed disease, at dose levels ≥120 μg/kg, 18 patients, or 45.0% of patients, achieved a complete response and another eight patients, or 20.0% of patients, achieved a partial response, resulting in a 65.0% ORR. |
| • | Lonca’s favorable clinical activity was observed across a broad patient population in this clinical trial, including transplant eligible and ineligible patients, patients who have not responded to first-line therapy or any prior therapy and patients with bulky disease, double-hit and triple-hit disease and transformed disease. The tables below show the effect of tumor characteristics, age and response to prior therapy on response rate data at dose levels ≥120 µg/kg. |
Tumor Characteristics | | | Overall Response Rate, responders/total (%) | |||
Bulky disease* | | | Absent | | | 52/109 (47.7) |
| | | Present | | | 4/18 (22.2), including 2/18 (11.1) CR | |
Double-hit or triple-hit disease** | | | Absent | | | 51/105 (48.6) |
| | | Present | | | 5/22 (22.7), including 3/22 (13.6) CR | |
Transformed disease*** | | | No | | | 40/96 (41.7) |
| | | Yes | | | 16/31 (51.6), including 9/31 (29.0) CR | |
Age | | | Overall Response Rate, responders/total (%) |
Less than 65 | | | 25/69 (36.2), including 16/69 (23.2) CR |
65-74 | | | 18/36 (50.0), including 11/36 (30.6) CR |
More than 74 | | | 13/22 (59.1), including 4/22 (18.2) CR |
Response to Prior Therapy | | | Overall Response Rate, responders/total (%) | |||
Response to first-line therapy | | | Complete response | | | 32/49 (65.3) |
| | | Partial response | | | 12/33 (36.4) | |
| | | Any response | | | 44/82 (53.7) | |
| | | No response | | | 12/44 (27.3) | |
Response to most recent therapy | | | Complete response | | | 16/21 (76.2) |
| | | Partial response | | | 9/24 (37.5) | |
| | | Any response | | | 25/45 (55.6) | |
| | | No response | | | 31/82 (37.8) | |
| • | Across all dose levels, the median DoR was not reached for patients who achieved a complete response (indicating that more than half of the patients continued to show a complete response as of their most recent assessment) and 2.86 months for patients who achieved a partial response, for an overall DoR of 4.47 months. At dose levels ≥120 µg/kg, the median DoR was not reached for patients who achieved a complete response (indicating that more than half of the patients continued to show a complete response as of their most recent assessment) and was 2.69 months for patients who achieved a partial response, for an overall DoR of 4.17 months. The figure below shows the DoR by response at dose levels ≥120 µg/kg. |
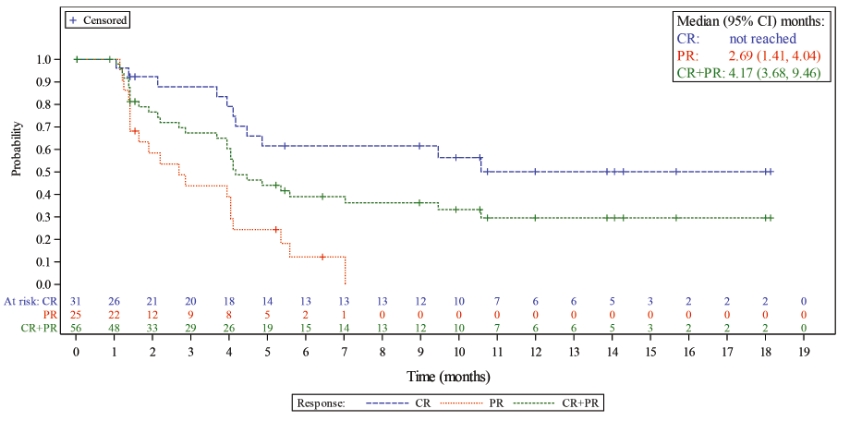
| • | In our post-hoc subset analysis that excluded patients with double-hit or triple-hit disease and patients with primary refractory and early relapsed disease, at dose levels ≥120 μg/kg, the median DoR was 10.6 months for patients who achieved a complete response and was 2.1 months for patients who achieved a partial response, for an overall DoR of 4.1 months. |
| • | Across all dose levels, five patients, or 33.3% of patients, achieved a complete response and another two patients, or 13.3% of patients, achieved a partial response, resulting in a 46.7% ORR. |
| • | The median DoR was not reached (indicating that more than half of the patients continued to show a complete response as of their most recent assessment). |
| • | Across all dose levels, nine patients, or 64.3% of patients, achieved a complete response and another two patients, or 14.3% of patients, achieved a partial response, resulting in a 78.6% ORR. |
| • | The median DoR was not reached (indicating that more than half of the patients continued to show a complete response as of their most recent assessment). |
Patient Characteristics | | | | | | | |||
Age, median (minimum, maximum) | | | 66 | | | (23, 94) | |||
Cancer characteristic, n (%) | | | Double-hit or triple-hit disease* | | | 13 | | | (9.0) |
| | | Transformed disease** | | | 26 | | | (17.9) | |
Number of previous chemotherapies received, median (minimum, maximum) | | | 3 | | | (2, 7) | |||
Response to first-line chemotherapy, n (%) | | | Relapsed | | | 99 | | | (68.3) |
| | | Refractory | | | 29 | | | (20.0) | |
Response to last-line chemotherapy, n (%) | | | Relapsed | | | 43 | | | (29.7) |
| | | Refractory | | | 82 | | | (56.6) | |
Prior stem cell transplant, n (%) | | | Yes | | | 24 | | | (16.6) |
| | | No | | | 121 | | | (83.4) | |
| • | With respect to the 145-patient interim analysis population, Grade ≥3 TEAEs were reported in 92 patients, or 63.4% of patients. The table below presents the most common Grade ≥3 TEAEs that were reported in more than 5% of patients. |
Grade ≥3 TEAEs*, n (%) | | | 52-Patient Futility Analysis Cohort (n=52) | | | 145-Patient Interim Analysis Population (n=145) | ||||||
Neutrophil count decreased** | | | 17 | | | (32.7) | | | 34 | | | (23.4) |
Gamma-glutamyltransferase increased | | | 13 | | | (25.0) | | | 20 | | | (13.8) |
Platelet count decreased** | | | 11 | | | (21.2) | | | 20 | | | (13.8) |
Anemia | | | 6 | | | (11.5) | | | 14 | | | (9.7) |
Hypercalcemia | | | 4 | | | (7.7) | | | *** | |||
Alanine aminotransferase increased | | | 3 | | | (5.8) | | | *** | |||
Hypokalemia | | | 3 | | | (5.8) | | | *** | |||
Leukopenia | | | 3 | | | (5.8) | | | *** | |||
Lymphocyte count decreased | | | 3 | | | (5.8) | | | 8 | | | (5.5) |
White blood cell count decreased | | | 3 | | | (5.8) | | | 8 | | | (5.5) |
Patients with any Grade ≥3 TEAEs | | | 41 | | | (78.8) | | | 92 | | | (63.4) |
| • | With respect to the 52-patient futility analysis cohort, TEAEs in 13 patients, or 25.0% of patients, led to treatment discontinuation, treatment-related adverse events in seven patients, or 13.5% of patients, led to treatment discontinuation and adverse events led to dose reductions in two patients, or 3.8% of patients. With respect to the 145-patient interim analysis population, TEAEs in 24 patients, or 16.6% of patients, led to treatment discontinuation, treatment-related adverse events in 13 patients, or 9.0% of patients, led to treatment discontinuation and adverse events led to dose reductions in 10 patients, or 6.9% of patients. |
| • | As of April 2020, 14 patients, or 21.2% of patients, who displayed a response have each tolerated ten or more cycles of treatment. |
| • | With respect to the 145-patient interim analysis population, 29 patients, or 20.0% of patients, achieved a complete response and another 37 patients, or 25.5% of patients, achieved a partial response, resulting in a 45.5% ORR. Six of the ten patients in the 52-patient futility analysis cohort who achieved a complete response later received stem cell transplant. The table below shows the response rate data. |
Best Overall Response, n (%) | | | 52-Patient Futility Analysis Cohort (n=52) | | | 145-Patient Interim Analysis Population (n=145) |
Complete response (CR) | | | 10 (19.2) | | | 29 (20.0) |
Partial response (PR) | | | 14 (26.9) | | | 37 (25.5) |
Stable disease | | | 10 (19.2) | | | 22 (15.2) |
Progressive disease | | | 10 (19.2) | | | 29 (20.0) |
Not evaluable | | | 8 (15.4) | | | 28 (19.3) |
Overall response rate (CR + PR) | | | 24 (46.2) | | | 66 (45.5) |
| • | Lonca’s favorable clinical activity was observed across a broad patient population in this clinical trial, including transplant eligible and ineligible patients, patients who have not responded to first-line therapy or any prior therapy, patients with bulky disease, double-hit and triple-hit disease and transformed disease and patients who have received prior CD19 therapies and SCT. The tables below show the effect of tumor characteristics, age, response to prior therapy and prior therapy (i.e., stem cell transplant or CAR-T) on response rate data. |
Tumor Characteristics | | | Overall Response Rate, responders/total (%) | |||
Bulky disease* | | | Absent | | | 64/136 (47.1) |
| | | Present | | | 2/9 (22.2), including 1/9 (11.1) CR | |
Double-hit or triple-hit disease** | | | Absent | | | 63/132 (47.7) |
| | | Present | | | 3/13 (23.1), including 3/13 (23.1) CR | |
Transformed disease*** | | | No | | | 54/119 (45.4) |
| | | Yes | | | 12/26 (46.2), including 6/26 (23.1) CR | |
Age | | | Overall Response Rate, responders/total (%) |
Less than 65 | | | 32/65 (49.2), including 10/65 (15.4) CR |
More than or equal to 65 | | | 34/80 (42.5), including 19/80 (23.8) CR |
Response to Prior Therapy | | | Overall Response Rate, responders/total (%) | |||
Response to first-line therapy | | | Refractory | | | 11/29 (37.9), including 3/29 (10.3) CR |
| | | Relapsed | | | 49/99 (49.5), including 22/99 (22.2) CR | |
Response to most recent therapy | | | Refractory | | | 28/82 (34.1), including 6/82 (7.3) CR |
| | | Relapsed | | | 28/43 (65.1), including 17/43 (39.5) CR | |
Response to any prior line therapy | | | Refractory | | | 8/22 (36.4), including 2/22 (9.1) CR |
| | | Relapsed | | | 57/117 (48.7), including 26/117 (22.2) CR | |
Prior Therapy | | | Overall Response Rate, responders/total (%) | |||
Stem cell transplant | | | Yes | | | 14/24 (58.3), including 7/24 (29.2) CR |
| | | No | | | 52/121 (43.0), including 22/121 (18.2) CR | |
CAR-T | | | Yes | | | 6/13 (46.2), including 2/13 (15.4) CR |
| | | No | | | 60/132 (45.5), including 27/132 (20.5) CR | |
| • | For the 52-patient futility analysis cohort, the median DoR was not reached for patients who achieved a complete response (indicating that more than half of the patients continued to show a complete response as of their most recent assessment) and was 5.68 months for patients who achieved a partial response, for an overall DoR of 6.87 months. The figure below shows the DoR by response for the 52-patient futility analysis cohort. |
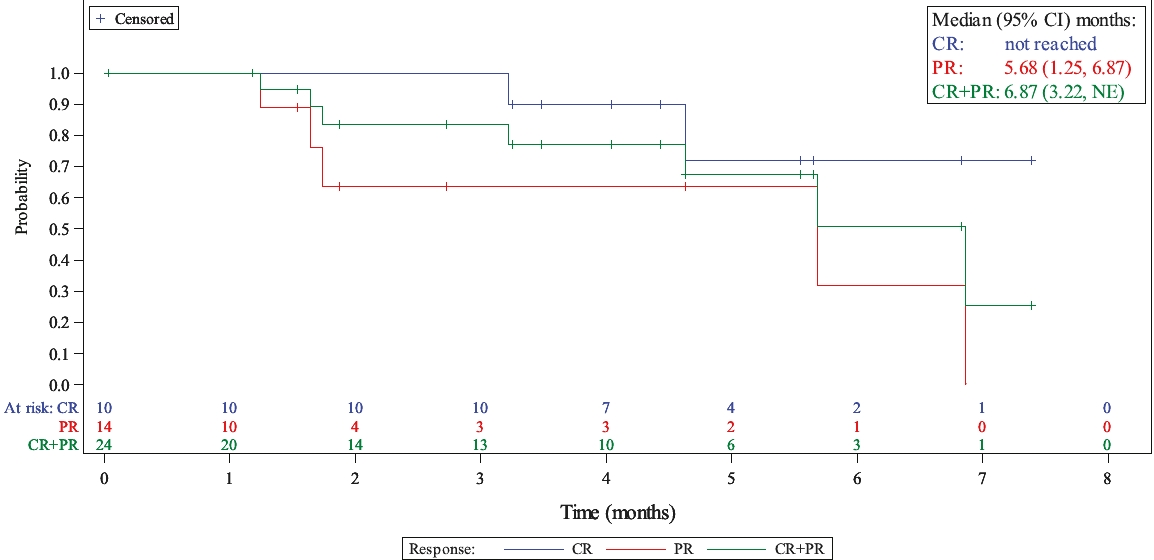
| • | We retain worldwide development and commercialization rights to Cami, subject to our collaboration and license agreement with Genmab. |
| • | Cami is being evaluated in a 100-patient pivotal Phase 2 clinical trial for the treatment of relapsed or refractory HL, for which we anticipate reporting top-line response rate data in the first half of 2021. |
| ○ | As of April 15, 2020, 47 patients have been enrolled in this pivotal Phase 2 clinical trial, which is currently subject to a partial clinical hold. See “Business—Camidanlumab Tesirine (ADCT-301): PBD-Based ADC Targeting CD25—Pivotal Phase 2 Clinical Trial in Relapsed or Refractory Hodgkin Lymphoma.” |
| • | We are advancing Cami through clinical development to support a BLA submission for the treatment of relapsed or refractory HL. |
| • | Enrollment has been completed in a 133-patient Phase 1 clinical trial of Cami for the treatment of relapsed or refractory HL and NHL, including 77 patients with relapsed or refractory HL. In this clinical trial, Cami demonstrated significant clinical activity across a broad patient population and maintained a tolerability profile that we believe was manageable. More specifically, as of April 2019, we observed: |
| ○ | At the initial dose for our pivotal Phase 2 clinical trial, an 86.5% ORR in heavily pre-treated patients with relapsed or refractory HL who have received a median of five prior lines of therapy, including patients who were relapsed or refractory to any or all of brentuximab vedotin, checkpoint inhibitors and stem cell transplant; and |
| ○ | A 44.0% ORR in heavily pre-treated patients with relapsed or refractory T-cell lymphoma who have received a median of four prior lines of therapy. |
| • | Cami is also being evaluated in a Phase 1b clinical trial for the treatment of selected advanced solid tumors by targeting Tregs, for which we anticipate reporting interim data at a medical conference in the second half of 2020. |
| ○ | In paired biopsies from three patients in the Phase 1b clinical trial, we have observed a significant increase in the ratio of T effector cells (“Teffs”) to Tregs. |
| • | At the initial dose for our pivotal Phase 2 clinical trial, an 86.5% ORR in heavily pre-treated patients with relapsed or refractory HL who were relapsed or refractory to any or all of brentuximab vedotin, checkpoint inhibitors and stem cell transplant; |
| • | Tolerability profile that we believe is manageable; |
| • | The potential opportunity to advance Cami into earlier lines of therapy as a monotherapy or in combination with other therapies; |
| • | Novel immuno-oncology approach targeting Tregs for the treatment of various advanced solid tumors; and |
| • | Convenient 30-minute intravenous infusions once every three weeks in the out-patient setting. |
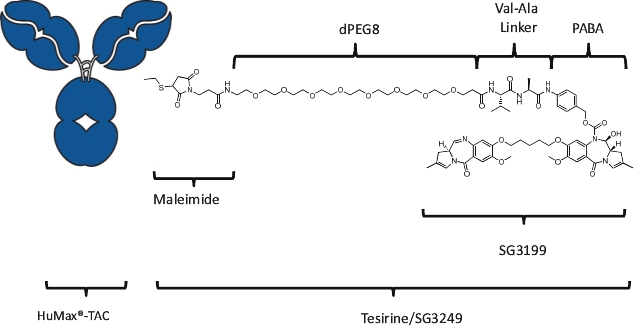
| • | CD25 expression in healthy human tissue is mainly limited to activated T cells and activated B cells. |
| • | CD25 is expressed in a wide range of hematological malignancies. |
| • | The importance of CD25 overexpression as a prognosticator in hematological malignancies has been shown in multiple indications, including DLBCL. |
| • | CD25 positive Treg cells have been shown to play a role in undermining anti-tumor immune functions. |
| • | The safety profiles of monoclonal antibodies directed against CD25 have been well characterized. |
| • | Clinical proof of concept for treatment of CD25 positive malignancies has been established using radio-immunoconjugates and immunotoxins incorporating the anti-CD25 antibodies. |
Patient Characteristics | | | | | | | |||
Age, median (minimum, maximum) | | | 52 | | | (19, 88) | |||
Number of previous systemic therapies received, median (minimum, maximum) | | | 5 | | | (1, 15) | |||
Prior stem cell transplant, n (%) | | | Yes | | | 57 | | | (44.5) |
| | | No | | | 71 | | | (55.5) | |
Patient Characteristics of HL Patients | | | | | | | |||
Age, median (minimum, maximum) | | | 38 | | | (19, 80) | |||
Number of previous systemic therapies received, median (minimum, maximum) | | | 5 | | | (2, 15) | |||
Prior stem cell transplant, n (%) | | | Yes | | | 47 | | | (61.0) |
| | | Autologous stem cell transplant | | | 39 | | | (50.6) | |
| | | Allogeneic stem cell transplant | | | 3 | | | (3.9) | |
| | | Both autologous and allogeneic stem cell transplant | | | 5 | | | (6.5) | |
| | | No | | | 30 | | | (39.0) | |
| • | The MTD was not reached in the dose escalation stage. |
| • | Grade ≥3 TEAEs were reported in 51 patients, or 66.2% of patients. The table below presents the most common Grade ≥3 TEAEs that are reported in more than 5% of patients. |
Grade ≥3 TEAEs, n (%) | | | Dose Levels | ||||||||||||
| | ≤20 µg/kg (n=3) | | | 30 µg/kg (n=20) | | | 45 µg/kg (n=37) | | | ≥60 µg/kg (n=17) | | | All Doses (n=77) | ||
Gamma-glutamyltransferase increased | | | 1 (33.3) | | | 2 (10.0) | | | 3 (8.1) | | | 7 (41.2) | | | 13 (16.9) |
Maculopapular rash | | | 1 (33.3) | | | 2 (10.0) | | | 8 (21.6) | | | 2 (11.8) | | | 13 (16.9) |
Alanine aminotransferase increased | | | 0 (0.0) | | | 0 (0.0) | | | 3 (8.1) | | | 4 (23.5) | | | 7 (9.1) |
Anemia | | | 1 (33.3) | | | 2 (10.0) | | | 3 (8.1) | | | 0 (0.0) | | | 6 (7.8) |
Aspartate aminotransferase increased | | | 0 (0.0) | | | 0 (0.0) | | | 1 (2.7) | | | 4 (23.5) | | | 5 (6.5) |
Guillain–Barré syndrome/polyradiculopathy | | | 0 (0.0) | | | 1 (5.0) | | | 3 (8.1) | | | 1 (5.9) | | | 5 (6.5) |
Lipase increased | | | 0 (0.0) | | | 1 (5.0) | | | 3 (8.1) | | | 0 (0.0) | | | 4 (5.2) |
Patients with any Grade ≥3 TEAEs | | | 2 (66.7) | | | 12 (60.0) | | | 25 (67.6) | | | 12 (70.6) | | | 51 (66.2) |
| • | TEAEs in 20 patients, or 26.0% of patients, led to treatment discontinuation. |
| • | Across all dose levels, 30 patients, or 40.0% of patients, achieved a complete response and another 23 patients, or 30.7% of patients, achieved a partial response, resulting in a 70.7% ORR. At the 45 µg/kg dose level, 18 patients, or 48.6% of patients, achieved a complete response and another 14 patients, or 37.8% of patients achieved a partial response, resulting in an 86.5% ORR. The table below shows the response rate data from this clinical trial. |
Best Overall Response, n (%) | | | Dose Levels | ||||||||||||
| | ≤20 µg/kg (n=3) | | | 30 µg/kg (n=18) | | | 45 µg/kg (n=37) | | | ≥60 µg/kg (n=17) | | | All Doses (n=75) | ||
Complete response (CR) | | | 0 (0.0) | | | 5 (27.8) | | | 18 (48.6) | | | 7 (41.1) | | | 30 (40.0) |
Partial response (PR) | | | 1 (33.3) | | | 4 (22.2) | | | 14 (37.8) | | | 4 (23.5) | | | 23 (30.7) |
Stable disease | | | 1 (33.3) | | | 6 (33.3) | | | 0 (0.0) | | | 1 (5.9) | | | 8 (10.7) |
Progressive disease | | | 0 (0.0) | | | 2 (11.1) | | | 5 (13.5) | | | 4 (23.5) | | | 11 (14.7) |
Not evaluable | | | 1 (33.3) | | | 1 (5.6) | | | 0 (0.0) | | | 1 (5.9) | | | 3 (4.0) |
Overall response rate (CR + PR) | | | 1 (33.3) | | | 9 (50.0) | | | 32 (86.5) | | | 11 (64.7) | | | 53 (70.7) |
| • | Cami’s favorable clinical activity was observed across a broad patient population in this clinical trial, including elderly patients and patients who have failed SCT. The table below shows the effect of age, prior therapy and response to prior therapy on response rate data at the 45 µg/kg dose level. |
Age | | | Overall Response Rate, responders/total (%) |
Less than or equal to 55 | | | 25/28 (89.3), including 14/28 (50.0) CR |
More than 55 | | | 7/9 (77.8), including 4/9 (44.4) CR |
Prior Therapy | | | Overall Response Rate, responders/total (%) |
Brentuximab vedotin | | | 32/37 (86.5) |
Brentuximab vedotin and checkpoint inhibitor | | | 23/26 (88.5) |
Stem cell transplant | | | 16/18 (88.9) |
Brentuximab vedotin, checkpoint inhibitor and stem cell transplant | | | 13/14 (92.9) |
Response to Prior Therapy | | | | | Overall Response Rate, responders/total (%) | |
Response to first-line therapy | | | Refractory | | | 11/13 (84.6), including 6/13 (46.2) CR |
| | | Relapsed | | | 21/24 (87.5), including 12/24 (50.0) CR | |
Response to most recent therapy | | | Refractory | | | 22/25 (88.0), including 11/25 (44.0) CR |
| | | Relapsed | | | 8/10 (80.0), including 6/10 (60.0) CR |
| • | Across all dose levels, the median DoR was 8.1 months for patients who achieved a complete response and 5.1 months for patients who achieved a partial response, for an overall DoR of 6.4 months. At the 45 µg/kg dose level, the median DoR was 7.2 months for patients who achieved a complete response and 5.6 months for patients who achieved a partial response, for an overall DoR of 6.6 months. The figure below shows the DoR by response at the 45 µg/kg dose level. |
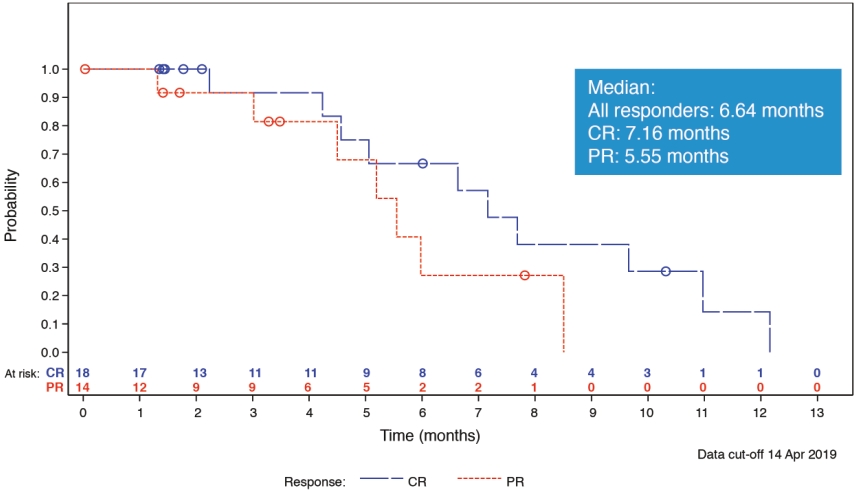
| • | The MTD was not reached in the dose escalation stage. |
| • | Grade ≥3 TEAEs were reported in 23 patients, or 79.3% of patients. The most common Grade ≥3 TEAEs, reported in more than 5% of patients, included hypercalcemia (10.3%), acute kidney injury (6.9%), back pain (6.9%), dehydration (6.9%), gamma-glutamyltransferase increased (6.9%), lung infection (6.9%), platelet count decreased (6.9%), pyrexia (6.9%), rash (6.9%) and maculopapular rash (6.9%). |
| • | TEAEs in two patients, or 6.9% of patients, led to treatment discontinuation. |
| • | Across all dose levels, two patients, or 8.0% of patients, achieved a complete response and another nine patients, or 36.0% of patients, achieved a partial response, resulting in a 44.0% ORR. |
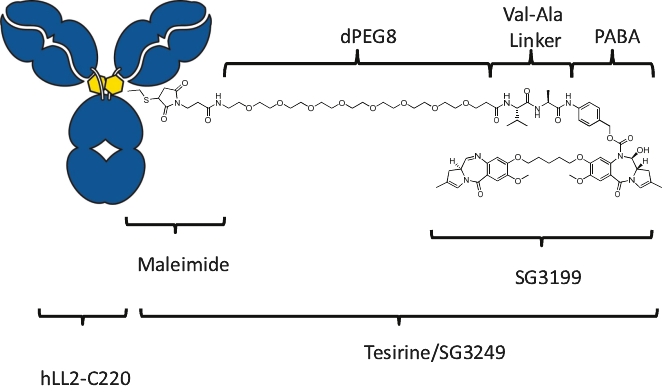
| • | The CD22 antigen is rapidly internalized by the cell. |
| • | An increasing number of reports describe the outgrowth of CD19-negative tumor cells in patients who initially respond to CD19-targeted therapy. We believe that given CD22’s broad and favorable expression profile, it may be a viable alternative B cell marker to CD19 for the targeted delivery of highly potent cytotoxic drugs. |
| | | n (%) | ||||||||||
Response | | | ADCT-602 0.3 mg/kg (n=10) | | | ADCT-602 1 mg/kg (n=10) | | | Non-Targeted ADC 1 mg/kg (n=10) | | | Vehicle Control (n=10) |
Complete response | | | 0 (0.0) | | | 10 (100.0) | | | 0 (0.0) | | | 0 (0.0) |
Partial response | | | 0 (0.0) | | | 0 (0.0) | | | 0 (0.0) | | | 0 (0.0) |
Tumor-free survivor | | | 0 (0.0) | | | 9 (90.0) | | | 0 (0.0) | | | 0 (0.0) |
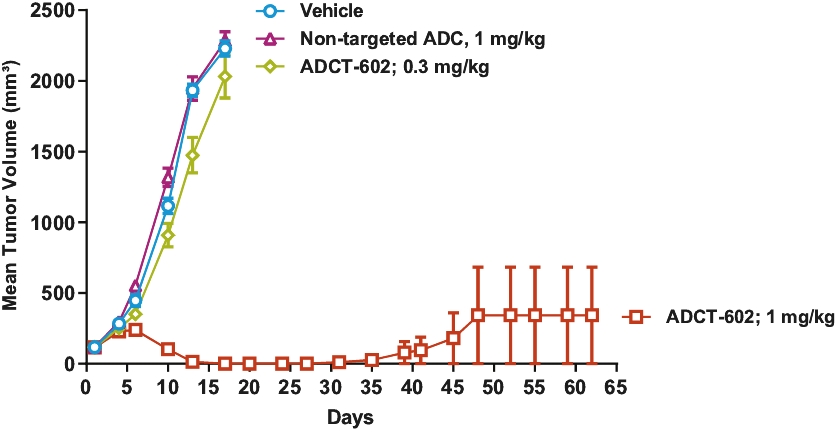
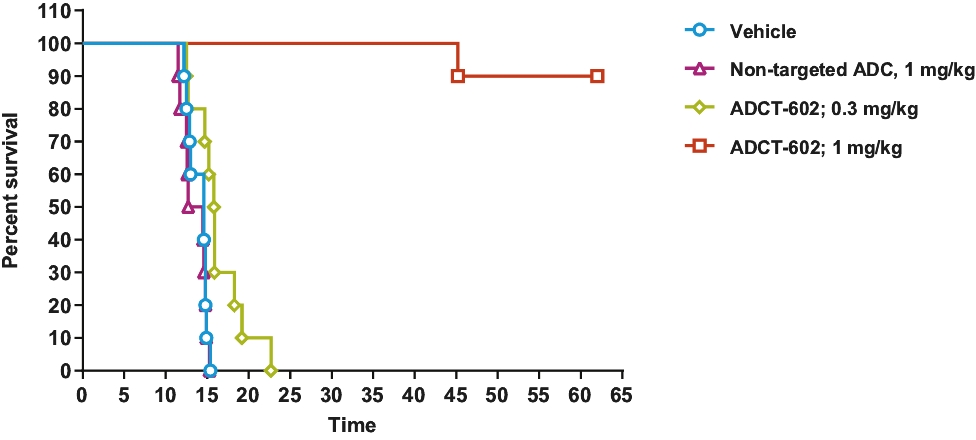

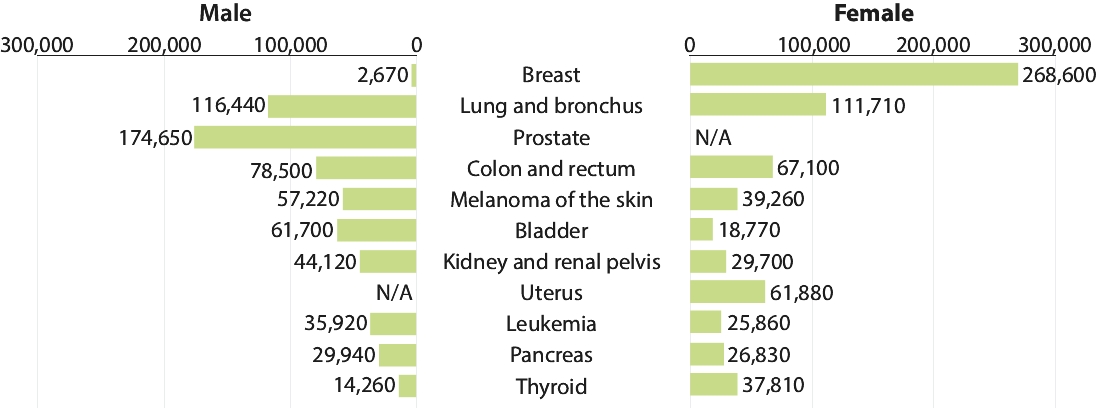

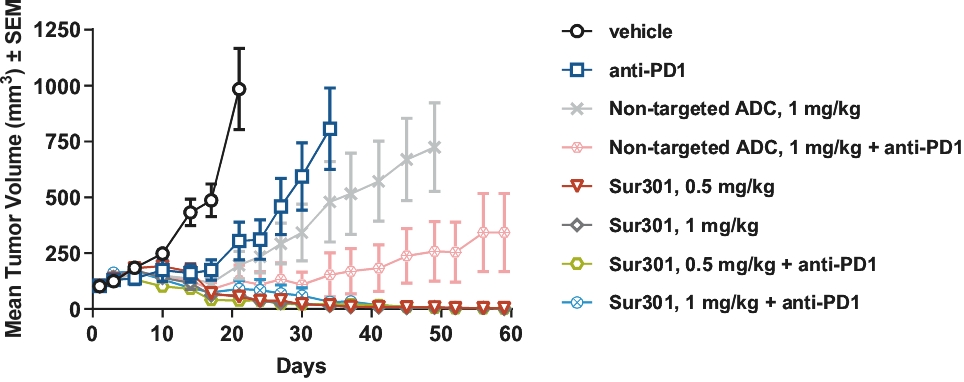
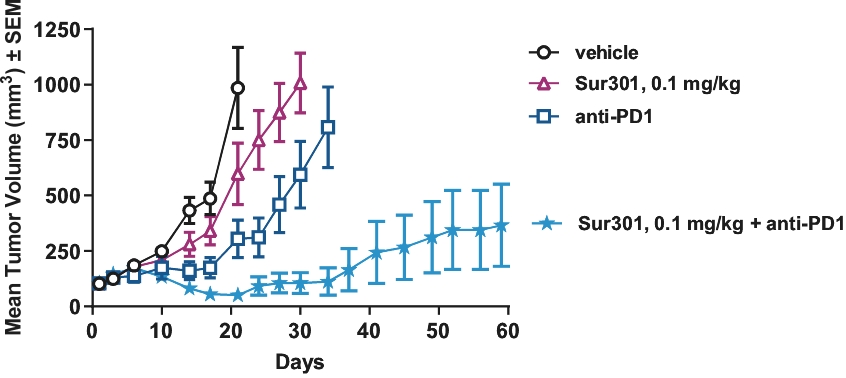
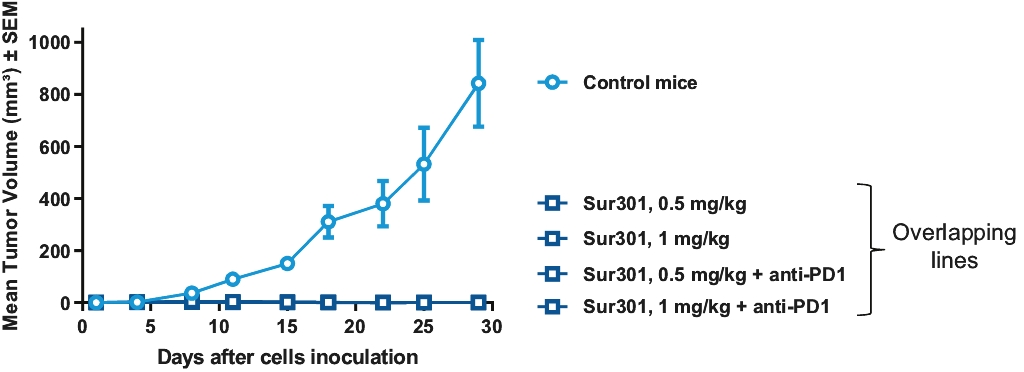
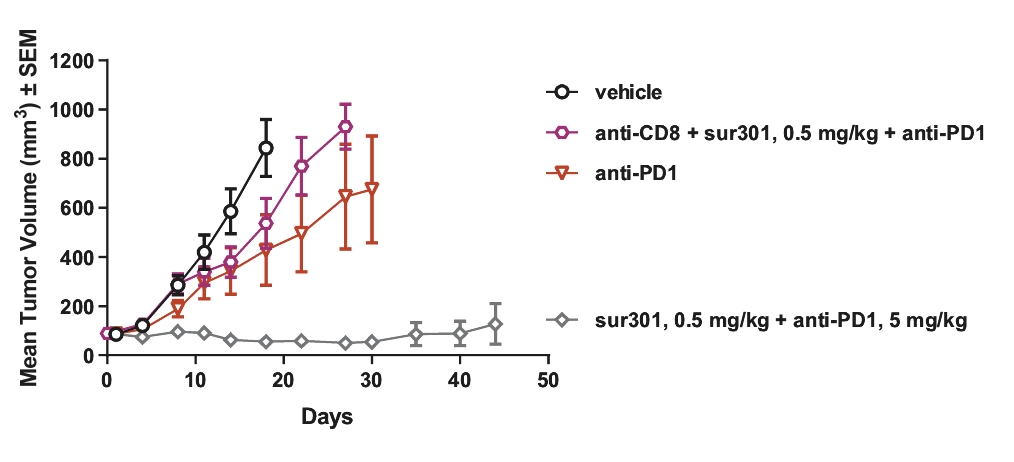
| • | Sur301 exhibited strong and durable anti-tumor activity that is superior to that achieved by an anti-PD1 antibody; |
| • | Sur301 exhibited strong synergistic effects when tested at a low dose level in combination with an anti-PD1 regimen; |
| • | Sur301’s observed anti-tumor activity was dependent on CD8 Teffs, and a statistically significant increase in the ratio of intratumoral CD8+ Teffs to Tregs was observed after administration of Sur301; and |
| • | Sur301 was associated with immunological memory in our re-challenge of tumor-free survivors. |
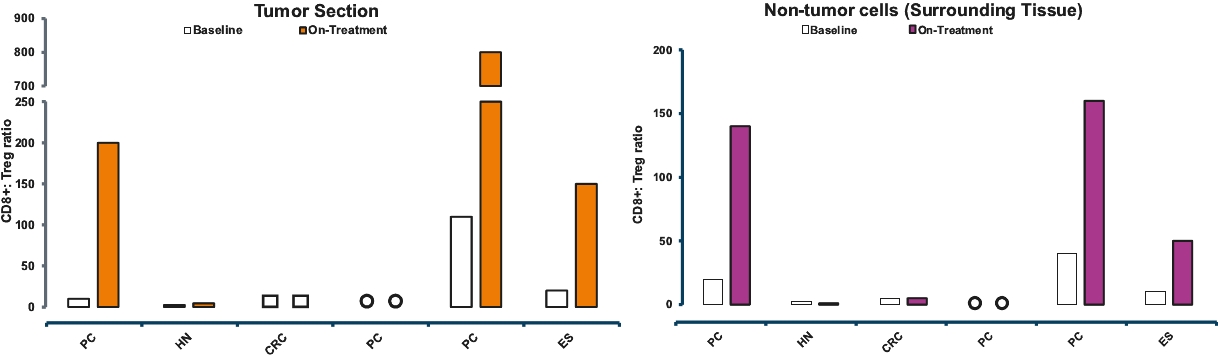
| • | ADCT-601 uses GlycoconnectTM site-specific conjugation technology, which allows for fast and stable conjugation of the warhead to the antibody. |
| • | The PBD payload of ADCT-601 contains a unique spacer, HydraspaceTM, which we have shown to provide an additional improvement in therapeutic index in preclinical models. |
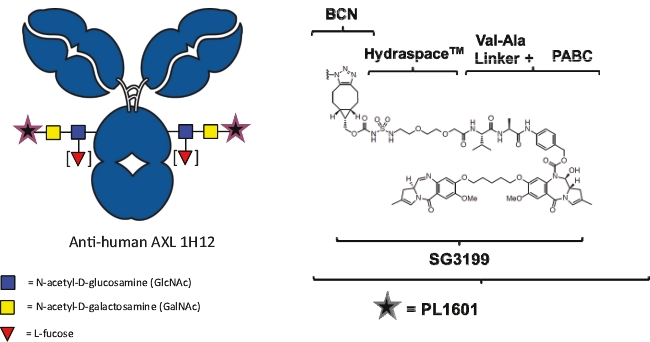
| • | AXL is highly overexpressed or ectopically expressed in a multitude of solid tumors, including in lung, breast, prostate, pancreas, glioma and esophageal cancers. Its overexpression is maintained in both primary tumors and metastasis. |
| • | AXL expression in healthy tissues is significantly lower than that in tumor cells. |
| • | AXL is expressed on M2 macrophages, which are part of the immunosuppressive tumor microenvironment. |
| • | Expression and activation of AXL is associated with poor clinical prognosis in many tumor indications and several studies suggest that expression of AXL is induced by both targeted and chemotherapy drugs. Therefore, AXL-based therapies may be efficacious even where traditional therapies have failed. |
| • | AXL is prevalent in tumors resistant to anti-PD1 therapy, and pre-clinical data have shown the benefit of combining AXL-targeted therapies with immunotherapies. |
| • | The extracellular portion of AXL can be cleaved off from the membrane to generate soluble AXL (“sAXL”), which can be detected in serum. Recent studies suggest that sAXL can be a potential circulating biomarker in certain tumors, representing a potentially attractive biomarker for clinical use. |
Response | | | n (%) | ||||||
| | ADCT-601 1 mg/kg (n=10) | | | Non-Targeted ADC 1 mg/kg (n=10) | | | Vehicle Control (n=10) | ||
Complete response | | | 4 (40.0) | | | 0 (0.0) | | | 0 (0.0) |
Partial response | | | 5 (50.0) | | | 0 (0.0) | | | 0 (0.0) |
Tumor-free survivor | | | 4 (40.0) | | | 0 (0.0) | | | 0 (0.0) |
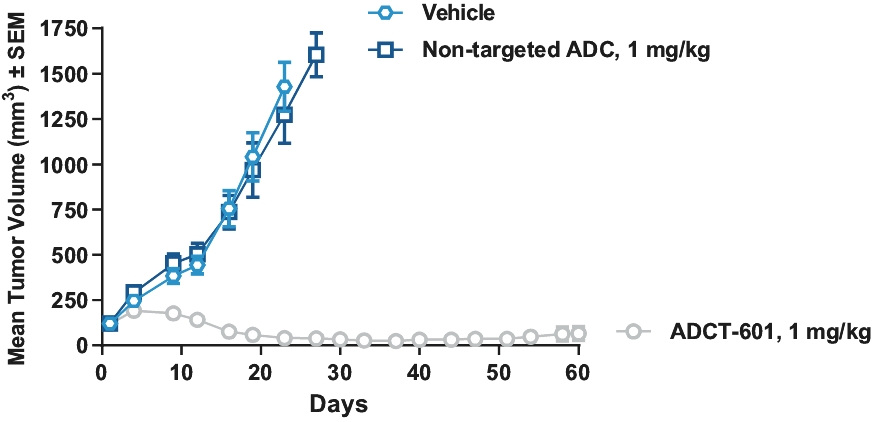
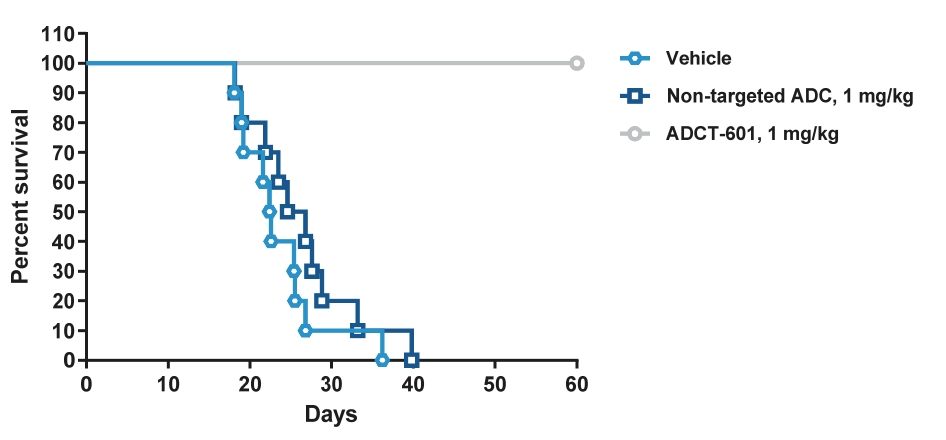
Response | | | n (%) | ||||||
| | ADCT-601 1 mg/kg (n=8) | | | Non-Targeted ADC 1 mg/kg (n=8) | | | Vehicle Control (n=8) | ||
Complete response | | | 2 (25.0) | | | 0 (0.0) | | | 0 (0.0) |
Partial response | | | 5 (62.5) | | | 0 (0.0) | | | 0 (0.0) |
Tumor-free survivor | | | 2 (25.0) | | | 0 (0.0) | | | 0 (0.0) |
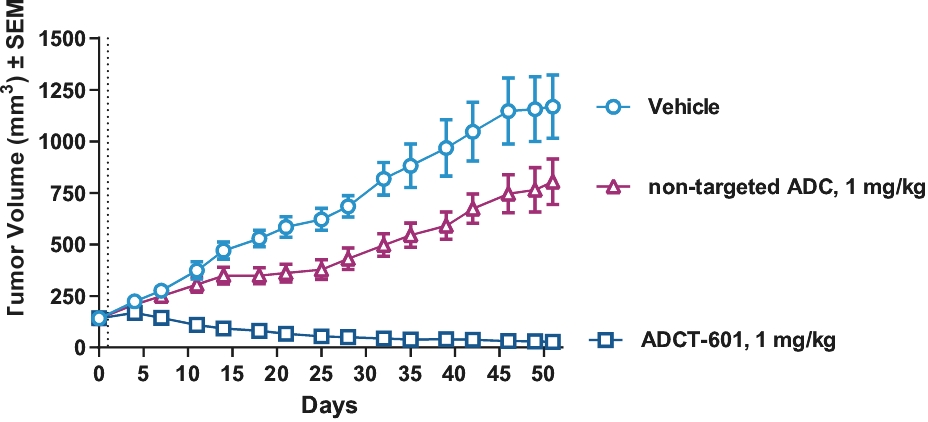
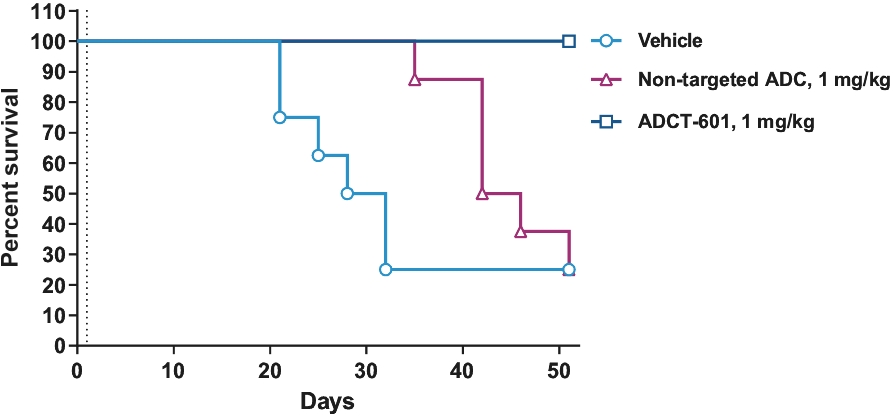
| | | n (%) | ||||||||||||||||
Response | | | ADCT-601 0.075 mg/kg (n=8) | | | ADCT-601 0.15 mg/kg (n=8) | | | ADCT-601 0.3 mg/kg (n=8) | | | AXL-107-MMAE 0.3 mg/kg (n=8) | | | AXL-107-MMAE 4 mg/kg (n=8) | | | Vehicle Control (n=8) |
Complete response | | | 0 (0.0) | | | 0 (0.0) | | | 3 (37.5) | | | 0 (0.0) | | | 8 (100.0) | | | 0 (0.0) |
Partial response | | | 0 (0.0) | | | 0 (0.0) | | | 1 (12.5) | | | 0 (0.0) | | | 0 (0.0) | | | 0 (0.0) |
Tumor-free survivor | | | 0 (0.0) | | | 0 (0.0) | | | 3 (37.5) | | | 0 (0.0) | | | 8 (100.0) | | | 0 (0.0) |
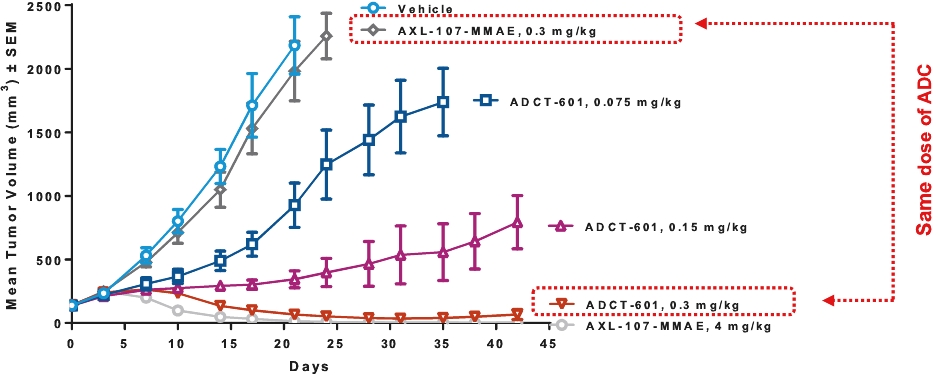

| | | n (%) | |||||||
Response | | | ADCT-901 1 mg/kg (n=8) | | | Non-Targeted ADC 1 mg/kg (n=8) | | | Vehicle Control (n=8) |
Complete response | | | 0 (0.0) | | | 0 (0.0) | | | 0 (0.0) |
Partial response | | | 7 (87.5) | | | 0 (0.0) | | | 0 (0.0) |
Tumor-free survivor | | | 0 (0.0) | | | 0 (0.0) | | | 0 (0.0) |
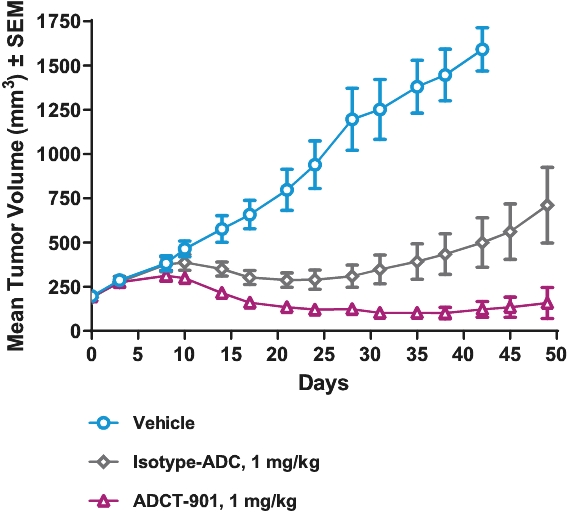
| | | n (%) | |||||||||||||
Response | | | ADCT-701 0.1 mg/kg (n=8) | | | ADCT-701 0.3 mg/kg (n=8) | | | ADCT-701 1 mg/kg (n=8) | | | Non-Targeted ADC 1 mg/kg (n=8) | | | Vehicle Control (n=8) |
Complete response | | | 0 (0.0) | | | 0 (0.0) | | | 2 (37.5) | | | 0 (0.0) | | | 0 (0.0) |
Partial response | | | 0 (0.0) | | | 0 (0.0) | | | 3 (62.5) | | | 0 (0.0) | | | 0 (0.0) |
Tumor-free survivor | | | 0 (0.0) | | | 0 (0.0) | | | 0 (0.0) | | | 0 (0.0) | | | 0 (0.0) |
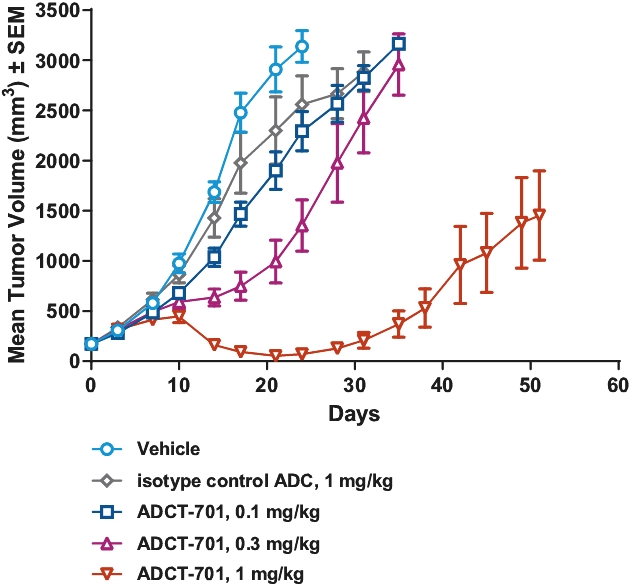
| • | completion of extensive preclinical laboratory and animal studies in accordance with applicable regulations, including studies conducted in accordance with GLP requirements; |
| • | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| • | approval by an IRB or independent ethics committee at each clinical trial site before each clinical trial may be commenced; |
| • | performance of adequate and well-controlled human clinical trials in accordance with applicable IND regulations, GCP requirements and other clinical trial-related regulations to establish the safety and efficacy of the investigational product for each proposed indication; |
| • | submission to the FDA of a BLA; |
| • | a determination by the FDA within 60 days of its receipt of a BLA to accept the filing for review; |
| • | satisfactory completion of one or more FDA pre-approval inspections of the manufacturing facility or facilities where the biologic, or components thereof, will be produced to assess compliance with cGMP requirements to assure that the facilities, methods and controls are adequate to preserve the biologic’s identity, strength, quality and purity; |
| • | satisfactory completion of any potential FDA audits of the clinical trial sites that generated the data in support of the BLA to assure compliance with GCPs and integrity of the clinical data; |
| • | payment of any user fees for FDA review of the BLA; |
| • | FDA review and approval of the BLA, including consideration of the views of any FDA advisory committee; and |
| • | compliance with any post-approval requirements, including REMS, where applicable, and post-approval studies required by the FDA as a condition of approval. |
| • | Phase 1 clinical trials generally involve a small number of healthy volunteers or disease-affected patients who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacokinetics, pharmacologic action, side effect tolerability, safety of the product candidate, and, if possible, early evidence of effectiveness. Phase 1 clinical trials may be designated as Phase 1a, which may involve dose escalation to determine the maximum tolerated dose, or Phase 1b, which may involve dose expansion at one or more dose levels to determine the recommended dose level for Phase 2 clinical trials. |
| • | Phase 2 clinical trials generally involve studies in disease-affected patients to evaluate proof of concept and/or determine the dosing regimen(s) for subsequent investigations. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, possible adverse effects and safety risks are identified, and a preliminary evaluation of efficacy is conducted. |
| • | Phase 3 clinical trials generally involve a large number of patients at multiple sites and are designed to provide the data necessary to demonstrate the effectiveness of the product for its intended use, its |
| • | restrictions on the marketing or manufacturing of the product, suspension of the approval, complete withdrawal of the product from the market or product recalls; |
| • | fines, warning or other enforcement-related letters or holds on post-approval clinical studies; |
| • | refusal of the FDA to approve pending BLAs or supplements to approved BLAs, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; or |
| • | injunctions or the imposition of civil or criminal penalties. |
| • | preclinical laboratory tests, animal studies and formulation studies all performed in accordance with the applicable EU Good Laboratory Practice regulations; |
| • | submission to the relevant national authorities of a clinical trial application (“CTA”) for each trial in humans, which must be approved before the trial may begin in each country where patient enrollment is planned; |
| • | performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product for each proposed indication; |
| • | submission to the relevant competent authorities of a MAA, which includes the data supporting safety and efficacy as well as detailed information on the manufacture and composition of the product in clinical development and proposed labelling; |
| • | satisfactory completion of an inspection by the relevant national authorities of the manufacturing facility or facilities, including those of third parties, at which the product is produced to assess compliance with strictly enforced cGMP; |
| • | potential audits of the non-clinical and clinical trial sites that generated the data in support of the MAA; and |
| • | review and approval by the relevant competent authority of the MAA before any commercial marketing, sale or shipment of the product. |
| • | The federal Anti-Kickback Statute, which prohibits any person or entity from, among other things, knowingly and willfully soliciting, receiving, offering or paying any remuneration, directly or indirectly, |
| • | Federal civil and criminal false claims laws, such as the FCA, which can be enforced by private citizens through civil qui tam actions, and civil monetary penalty laws prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented, false, fictitious or fraudulent claims for payment of federal funds, and knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to avoid, decrease or conceal an obligation to pay money to the federal government. For example, pharmaceutical companies have been prosecuted under the FCA in connection with their alleged off-label promotion of drugs, purportedly concealing price concessions in the pricing information submitted to the government for government price reporting purposes, and allegedly providing free product to customers with the expectation that the customers would bill federal healthcare programs for the product. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes “any request or demand” for money or property presented to the U.S. government. In addition, manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. |
| • | HIPAA, among other things, imposes criminal liability for executing or attempting to execute a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and creates federal criminal laws that prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services. |
| • | HIPAA, as amended by HITECH, and their implementing regulations, which impose privacy, security and breach reporting obligations with respect to individually identifiable health information upon entities subject to the law, such as health plans, healthcare clearinghouses and certain healthcare providers, known as covered entities, and their respective business associates that perform services for them that involve individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in U.S. federal courts to enforce HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. |
| • | Federal and state consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers. |
| • | The federal transparency requirements under the Physician Payments Sunshine Act, created under the Health Care Reform Act, which requires, among other things, certain manufacturers of drugs, devices, biologics and medical supplies reimbursed under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to CMS information related to payments and other transfers of value provided to physicians, as defined by such law, and teaching hospitals and physician ownership and investment interests, including such ownership and investment interests held by a physician’s immediate family members. |
| • | State and foreign laws that are analogous to each of the above federal laws, such as anti-kickback and false claims laws, that may impose similar or more prohibitive restrictions, and may apply to items or services reimbursed by non-governmental third-party payors, including private insurers. |
| • | State and foreign laws that require pharmaceutical companies to implement compliance programs, comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or to track and report gifts, compensation and other remuneration provided to physicians and other healthcare providers; state laws that require the reporting of marketing expenditures or drug pricing, including information pertaining to and justifying price increases; state and local laws that require the registration of pharmaceutical sales representatives; state laws that prohibit various marketing-related activities, such as the provision of certain kinds of gifts or meals; state laws that require the posting of information relating to clinical trials and their outcomes; and other federal, state and foreign laws that govern the privacy and security of health information or personally identifiable information in certain circumstances, including state health information privacy and data breach notification laws which govern the collection, use, disclosure, and protection of health-related and other personal information, many of which differ from each other in significant ways and often are not pre-empted by HIPAA, thus requiring additional compliance efforts. |
| • | increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program; |
| • | established a branded prescription drug fee that pharmaceutical manufacturers of certain branded prescription drugs must pay to the federal government; |
| • | expanded the list of covered entities eligible to participate in the 340B drug pricing program by adding new entities to the program; |
| • | established a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| • | extended manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| • | expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| • | created a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for certain drugs and biologics, including our product candidates, that are inhaled, infused, instilled, implanted or injected; |
| • | established a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; |
| • | established a Center for Medicare and Medicaid Innovation at the CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending; and |
| • | created a licensure framework for follow-on biologic products. |
| • | a covered benefit under its health plan; |
| • | safe, effective and medically necessary; |
| • | appropriate for the specific patient; |
| • | cost-effective; and |
| • | neither experimental nor investigational. |
Location | | | Primary Function | | | Approximate Size |
Biopôle Route de la Corniche 3B 1066 Epalinges Switzerland | | | Office | | | 518 m2 |
| | | | | |||
430 Mountain Avenue, 4th Floor Murray Hill, New Jersey 07974 United States | | | Office | | | 1038 m2 |
| | | | | |||
42 New Road London, E1 2AX United Kingdom | | | Office and laboratory | | | 284 m2 |
| | | | | |||
1510 Fashion Island Boulevard, Suite 205 San Mateo, California 94404 United States | | | Office | | | 390 m2 |
Name | | | Position(s) | | | Age |
Executive Officers and Directors | | | | | ||
Christopher Martin | | | Chief Executive Officer and Director | | | 61 |
Michael Forer | | | Executive Vice President, Chief Financial Officer and Vice Chairman of the Board of Directors(1) | | | 54 |
Jay Feingold | | | Senior Vice President, Chief Medical Officer & Head of Oncology | | | 63 |
Jennifer Herron | | | Senior Vice President, Chief Commercial Officer | | | 50 |
Patrick van Berkel | | | Senior Vice President, Research and Development | | | 51 |
Richard Onyett | | | Vice President, Business Development | | | 72 |
Dominique Graz | | | General Counsel and Company Secretary | | | 59 |
Peter Greaney | | | Head of Corporate Development | | | 40 |
Stéphane Henchoz | | | Director of Finance | | | 52 |
Susan Romanus | | | Chief Compliance Officer | | | 54 |
Jennifer Creel | | | Chief Financial Officer Nominee(1) | | | 49 |
Non-Executive Directors | | | | | ||
Ron Squarer | | | Chairman of the Board of Directors | | | 53 |
Peter B. Corr | | | Director | | | 72 |
Stephen Evans-Freke | | | Director | | | 68 |
Peter Hug | | | Director | | | 61 |
Thomas Pfisterer | | | Director | | | 38 |
Thomas M. Rinderknecht | | | Director | | | 66 |
Tyrell J. Rivers | | | Director | | | 47 |
Victor Sandor | | | Director | | | 53 |
Jacques Theurillat | | | Director | | | 60 |
| (1) | We intend for Ms. Creel to become our Chief Financial Officer upon the consummation of this offering or on June 1, 2020, whichever is earlier, after which time Mr. Forer will remain our Executive Vice President and Vice Chairman of our board of directors. |
| • | pre-approve the audit services and non-audit services (including the fees and terms thereof) to be provided by the independent auditor pursuant to pre-approval policies and procedures; |
| • | evaluate the independent auditor’s qualifications, performance and independence, and present its conclusions with respect to the independent auditor to the board of directors on at least an annual basis; |
| • | confirm and evaluate the rotation of the audit partners on the audit engagement team as required by law; |
| • | at least annually, review management’s plans with respect to the responsibilities, budget and staffing of the internal audit function and its plans for the implementation of the internal audit function, if any; |
| • | review and discuss with management and the independent auditor the annual audited consolidated and stand-alone financial statements and unaudited quarterly financial statements; |
| • | review with management, personnel responsible for the design and implementation of the internal audit function and the independent auditor (i) any analyses or other written communications prepared by management and/or the independent auditor setting forth significant financial reporting issues and judgments made in connection with the preparation of the financial statements, (ii) the Company’s critical accounting policies and practices, (iii) the effect of regulatory and accounting initiatives, as well as off-balance sheet transactions and structures, on the Company’s financial statements and (iv) any major issues regarding accounting principles and financial statement presentations; |
| • | review the type and presentation of information included in the earnings press releases, as well as financial information and earnings guidance provided to analysts and rating agencies, and may review earnings press releases prior to public dissemination; |
| • | in conjunction with the chief executive officer and chief financial officer, review disclosure controls and procedures and internal control over financial reporting; |
| • | review policies and practices with respect to risk assessment and risk management; and |
| • | review any major litigation or investigations against the Company that may have a material impact on the Company’s financial statements. |
| • | regularly review and make recommendations to the board of directors regarding our compensation and benefits strategy and guidelines; |
| • | prepare the proposals to the shareholders’ meeting regarding the compensation of the members of the board of directors and the executive committee; |
| • | regularly review and make recommendations to the board of directors regarding the compensation of the members of the board of directors and of the executive committee; |
| • | review and approve the recommendation of our chief executive officer regarding the fixed and variable compensation, including incentive plan participation and benefits, of the members of the management team other than members of the executive committee; |
| • | review and make recommendations to the board of directors regarding our compensation and benefits plans (cash and/or equity-based plans) and, where appropriate or required, make recommendations to adopt, amend and terminate such plans; |
| • | to the extent not delegated by the compensation committee to a different body or a third party, administer our compensation and benefits plans (other than equity-based plans); and |
| • | review and assess risks arising from our employee compensation policies and practices and whether any such risks are reasonably likely to have a material adverse effect on us. |
| • | determine selection criteria for the succession of the members of the board of directors and board committees, our chief executive officer, our chief financial officer and our executive vice president, and establish such succession planning (including for the event of the incapacitation, retirement or removal of such individuals) by making recommendations to the board of directors; |
| • | oversee searches and identify qualified individuals for membership on the board of directors and for the position of chief executive officer; |
| • | recommend individuals for membership on the board of directors and board committees and for the position of chief executive officer; |
| • | at least annually, prepare the board of directors’ assessment of the performance of the board of directors and board committees and of our chief executive officer and review the recommendations of the other board committees based on their evaluation of their own performance; |
| • | review the recommendations of the other board committees based on their self-evaluations and discuss its self-evaluation with the board of directors; |
| • | monitor and assess developments and trends in corporate governance to the extent that these do not have an impact on the activities and tasks of the audit and finance committee or the compensation committee; |
| • | review proposals to be made to the board of directors for the amendment of our amended and restated articles of association, our organizational regulations, any other rules or regulations and the Code of Conduct; |
| • | periodically review and reassess the adequacy of the Code of Conduct and recommend any proposed changes to the board of directors; |
| • | periodically review and assess the adequacy of the charter of the nomination and corporate governance committee and recommend any proposed changes to the board of directors for approval; |
| • | if it deems advisable, develop and recommend to the board of directors corporate governance guidelines for the Company, and, if such guidelines are adopted, periodically review and reassess the adequacy of such guidelines, consider any requests for waivers of such guidelines and make recommendations to the board of directors regarding amendments and requests for waivers; and |
| • | oversee compliance with the Code of Conduct and report on such compliance to the board of directors. |
| • | Exemption from the requirement that a majority of the board of directors be comprised of independent directors and that there be regularly scheduled meetings with only the independent directors present. Swiss law does not have such a requirement. |
| • | Exemption from the requirements that the compensation committee and the nomination and corporate governance committee be comprised of independent directors. Swiss law does not have such requirements. |
| • | Exemption from quorum requirements applicable to meetings of shareholders. Swiss law does not require such quorum requirements. |
| • | Exemption from the requirement that independent directors meet at regularly scheduled executive sessions. Swiss law does not have such a requirement. |
| • | Exemption from the requirement that listed companies adopt and disclose corporate governance guidelines that cover certain minimum specified subjects related to director qualifications and responsibilities. Swiss law does not require the adoption or disclosure of such guidelines. |
| • | Exemption from the requirement to disclose within four business days of any determination to grant a waiver of the Code of Conduct to directors and executive officers. Although we will require approval by our board of directors for any such waiver, we may choose not to disclose the waiver in the manner set forth in the NYSE listing standards. |
| • | Exemption from the requirement to obtain shareholder approval for certain issuances of securities, including shareholder approval of share option plans. Our amended and restated articles of association will provide that our board of directors is authorized, in certain instances, to issue a certain number of common shares without re-approval by our shareholders. |
| • | each person, or group of affiliated persons, known by us to own beneficially 5% or more of our outstanding common shares; |
| • | each of our executive officers and directors and persons nominated to serve in such positions; and |
| • | all executive officers and directors and persons nominated to serve in such positions as a group. |
| | | Number of Common Shares Beneficially Owned | | | Percentage of Common Shares Beneficially Owned | ||||
Principal Shareholders | | | Before This Offering | | | After This Offering | |||
5% Shareholders | | | | | | | |||
Entities affiliated with Auven Therapeutics GP Ltd.(1) | | | 23,198,730 | | | 41.0% | | | 36.3% |
AstraZeneca UK Limited(2) | | | 3,811,215 | | | 6.7% | | | 6.0% |
HPWH TH AG(3) | | | 6,000,000 | | | 10.6% | | | 9.4% |
Redmile Alpha LLC(4) | | | 3,210,770 | | | 5.7% | | | 5.0% |
Executive Officers and Directors | | | | | | | |||
Peter B. Corr(5) | | | 23,198,730 | | | 41.0% | | | 36.3% |
Jennifer Creel | | | * | | | * | | | * |
Stephen Evans-Freke(5) | | | 23,198,730 | | | 41.0% | | | 36.3% |
Jay Feingold | | | * | | | * | | | * |
Michael Forer(6) | | | 864,678 | | | 1.5% | | | 1.4% |
Dominique Graz | | | * | | | * | | | * |
Peter Greaney | | | * | | | * | | | * |
Stéphane Henchoz | | | * | | | * | | | * |
Jennifer Herron | | | * | | | * | | | * |
Peter Hug | | | * | | | * | | | * |
Christopher Martin(7) | | | 1,131,745 | | | 2.0% | | | 1.8% |
Richard Onyett | | | * | | | * | | | * |
Thomas Pfisterer | | | * | | | * | | | * |
Thomas M. Rinderknecht | | | * | | | * | | | * |
Tyrell J. Rivers(8) | | | * | | | * | | | * |
Susan Romanus | | | * | | | * | | | * |
Victor Sandor | | | * | | | * | | | * |
Ron Squarer | | | * | | | * | | | * |
Jacques Theurillat | | | * | | | * | | | * |
Patrick van Berkel(9) | | | * | | | * | | | * |
All executive officers and directors as a group (20 persons)(10) | | | 26,611,735 | | | 47.0% | | | 41.6% |
| * | Less than 1% of our total outstanding common shares. |
| (1) | Consists of (i) 17,878,048 common shares held of record by A.T. Holdings II Sàrl (“A.T. Holdings”), a Swiss limited liability company (société à responsabilité limitée) and (ii) 5,320,682 common shares held of record by ADC Products Switzerland Sàrl (“ADC Products”), a Swiss limited liability company (société à responsabilité limitée). A.T. Holdings is a wholly-owned subsidiary of C.T. Phinco Sàrl, a Swiss limited liability company (société à responsabilité limitée). C.T. Phinco Sàrl is a wholly-owned subsidiary of Auven Therapeutics Holdings L.P. (“Auven Therapeutics”). Auven Therapeutics GP Ltd. is the general partner of Auven Therapeutics General L.P. Auven Therapeutics General L.P. is the general partner of Auven Therapeutics. Auven Therapeutics holds a 73.8% ownership interest in ADC Products. As the sole shareholders of Auven Therapeutics GP Ltd., Mr. Corr and Mr. Evans-Freke may be deemed to have shared voting and investment power with respect to the common shares of ADCT. Although the managing directors of A.T. Holdings and ADC Products have voting rights with respect to the common shares held of record, the managing directors disclaim beneficial ownership over 23,198,730 of the common shares of ADCT. The number of common shares above does not reflect 3,999,193 common shares held of record by A.T. Holdings as nominee on behalf of equity incentive plan participants, for which A.T. Holdings takes into account the wishes of the equity incentive plan participants when exercising voting rights. 23,195,605 common shares of ADCT held of record by A.T. Holdings and ADC Products have been pledged pursuant to lending arrangements. The business address for Auven Therapeutics GP Ltd. is 171 Main Street, Road Town, Tortola, British Virgin Islands VG1110. |
| (2) | Consists of 3,811,215 common shares held of record by AstraZeneca UK Limited (“AstraZeneca”), a private limited company organized in the United Kingdom. AstraZeneca is a wholly-owned subsidiary of AstraZeneca PLC, a public limited company organized under the laws of the United Kingdom, which may be deemed to have sole voting and investment power. The registered office address of AstraZeneca is 1 Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge, United Kingdom, CB2 0AA. |
| (3) | Consists of 6,000,000 common shares held of record by HPWH TH AG (“HPWH”), a Swiss stock corporation. Dr. Hans-Peter Wild and Mr. Pfisterer are members of HPWH board of directors, and may be deemed to have shared voting and investment power with |
| (4) | Consists of 3,210,770 common shares held of record held by Redmile Alpha LLC (“Redmile Alpha”). Redmile Group, LLC (“Redmile”) is the managing member of Redmile Alpha and may be deemed to beneficially own the securities held by Redmile Alpha. Jeremy C. Green serves as the managing member of Redmile and as such shares voting and dispositive power over the shares held by Redmile Alpha. Redmile Group, LLC and Mr. Green each disclaim beneficial ownership of these shares, except to the extent of its or his pecuniary interest in such shares, if any. The address for Redmile Alpha, Redmile and Mr. Green is One Letterman Drive, Suite D3-300, San Francisco, California 94129. |
| (5) | Consists of (i) 17,878,048 common shares held of record by A.T. Holdings, a company controlled by Auven Therapeutics and (ii) 5,320,682 common shares held of record by ADC Products. Auven Therapeutics holds a 73.8% ownership interest in ADC Products. As described in footnote (1), the sole shareholders of Auven Therapeutics GP Ltd., Mr. Corr and Mr. Evans-Freke, may be deemed to have shared voting and investment power with respect to the common shares of ADCT. |
| (6) | Does not include 336,260 common shares held by Dune Capital Inc., a company which is wholly-owned by a trust whose beneficiaries include Mr. Forer and his family. Mr. Forer does not exercise investment or voting control over the trust, and therefore such shares do not appear in the table above. |
| (7) | Does not include 503,774.9 common shares held of record by Tuula Martin, spouse of Christopher Martin. Mr. Martin does not exercise investment or voting control over such shares, and therefore such shares do not appear in the table above. |
| (8) | Mr. Rivers, an executive director within AstraZeneca’s corporate development group, disclaims beneficial ownership with respect to the 3,811,215 common shares held of record by AstraZeneca. See footnote (2). |
| (9) | Consists of 229,806 common shares held by Betulamab B.V., a Dutch private limited liability company of which Dr. van Berkel is beneficial owner. The registered office address of Betulamab B.V. is Neerdyck 3, 3601 CZ Maarssen, The Netherlands. |
| (10) | Includes 1,304,650 common shares held by directors and executive officers who beneficially own less than 1.0% of shares and an estimated 111,932 common shares (subject to adjustments for final tax and social security deductions) subject to options that are immediately exercisable or exercisable within 60 days of the completion of this offering, which represent the common shares that will be issued to settle outstanding awards under our 2014 Incentive Plan in connection with this offering. |
Name of Shareholders | | | Number of Class E Preferred Shares (Number of Common Shares After Giving Effect to the Share Capital Reorganization and the Share Split) Purchased(1) | |||
A.T. Holdings II Sàrl | | | 105.0000 | | | (1,312,500.0000) |
ADC Products Switzerland Sàrl | | | 56.0000 | | | (700,000.0000) |
Michael Forer | | | 0.7709 | | | (9,636.2500) |
Tuula Martin(2) | | | 1.5418 | | | (19,272.5000) |
Thomas Rinderknecht | | | 3.3421 | | | (41,776.2500) |
Jacques Theurillat | | | 1.5815 | | | (19,768.7500) |
Barrie Ward(3) | | | 0.4667 | | | (5,833.7500) |
| (1) | Includes shares and fractional shares held by nominees on behalf of the shareholders set out in the table. |
| (2) | Tuula Martin is the spouse of Christopher Martin, who is a member of our board of directors and CEO. |
| (3) | Barrie Ward was a member of our board of directors from September 2014 to April 2020. |
Name of Shareholders | | | Number of Class E Preferred Shares (Number of Common Shares After Giving Effect to the Share Capital Reorganization and the Share Split) Purchased(1) | |||
A.T. Holdings II Sàrl(2) | | | 62.0000 | | | (775,000.0000) |
Michael Forer | | | 0.4286 | | | (5,357.5000) |
Dominique Graz | | | 0.5700 | | | (7,125.0000) |
Stéphane Henchoz | | | 0.1000 | | | (1,250.0000) |
Thomas Rinderknecht | | | 1.0000 | | | (12,500.0000) |
Jacques Theurillat | | | 0.1800 | | | (2,250.0000) |
Tuula Martin(3) | | | 0.4300 | | | (5,375.0000) |
Barrie Ward(4) | | | 0.0200 | | | (250.0000) |
| (1) | Includes shares and fractional shares held by nominees on behalf of the shareholders set out in the table. |
| (2) | Includes 29.0000 Class E preferred shares (which is equal to 362,500.0000 common shares after giving effect to the Share Capital Reorganization and the Share Split) originally acquired by ADC Products Switzerland Sàrl and later transferred to A.T. Holdings II Sàrl. |
| (3) | Tuula Martin is the spouse of Christopher Martin, who is a member of our board of directors and CEO. |
| (4) | Barrie Ward was a member of our board of directors from September 2014 to April 2020. |
Name of Shareholders | | | Number of Shares Transferred (After Giving Effect to the Conversion) |
ADC Products Switzerland Sàrl | | | 0.7727 |
Betulamab B.V.(1) | | | 0.4000 |
Michael Forer | | | 1.2801 |
Dominique Graz | | | 0.8000 |
Peter Greaney | | | 0.4000 |
Stéphane Henchoz | | | 0.9429 |
Peter Hug | | | 0.2000 |
Christopher Martin | | | 0.2000 |
Thomas Pfisterer | | | 0.2000 |
Thomas Rinderknecht | | | 1.5540 |
Jacques Theurillat | | | 1.1891 |
Tuula Martin(2) | | | 0.2458 |
| (1) | Betulamab B.V. is a Dutch private limited liability company of which Dr. van Berkel, who is our Senior Vice President, Research and Development, is beneficial owner. |
| (2) | Tuula Martin is the spouse of Christopher Martin, who is a member of our board of directors and CEO. |
Name of Director or Executive Officer | | | Interest Rate as of April 15, 2020 | | | Largest Amount Previously Outstanding Immediately Prior to Repayment (in USD thousands) |
Michael Forer | | | 2.25% | | | 3,307 |
Dominique Graz | | | 2.25% | | | 207 |
Peter Greaney | | | 2.25% | | | 71 |
Stéphane Henchoz | | | 2.25% | | | 336 |
Peter Hug | | | 2.25% | | | 229 |
Christopher Martin | | | 2.25% | | | 4,148 |
Thomas Pfisterer | | | 2.25% | | | 341 |
Thomas M. Rinderknecht | | | 2.25% | | | 472 |
Jacques Theurillat | | | 2.25% | | | 472 |
Patrick van Berkel | | | 2.25% | | | 472 |
Barrie Ward(1) | | | 2.25% | | | 385 |
| (1) | Barrie Ward was a member of our board of directors from September 2014 to April 2020. |
| • | On October 11, 2017, our share capital as registered with the Commercial Register on October 12, 2017, was increased by issuing 588 Class E preferred shares; |
| • | On October 24, 2017, our share capital as registered with the Commercial Register on October 30, 2017, was increased by issuing 22 Class E preferred shares; |
| • | On November 10, 2017, our share capital as registered with the Commercial Register on November 16, 2017, was increased by issuing 7 Class E preferred shares; |
| • | On June 19, 2018, our share capital as registered with the Commercial Register on June 29, 2018, was increased by issuing 3 Class A common shares; |
| • | On December 10, 2018, our share capital as registered with the Commercial Register on December 14, 2018, was increased by issuing 33 Class A common shares; |
| • | On January 30, 2019, our share capital as registered with the Commercial Register on February 6, 2019, was increased by issuing 6 Class A common shares; |
| • | On June 4, 2019, our share capital as registered with the Commercial Register on June 7, 2019, was increased by issuing 216 Class E preferred shares; |
| • | On June 7, 2019, our share capital as registered with the Commercial Register on June 14, 2019, was increased by issuing 2 Class E preferred shares; |
| • | On June 28, 2019, our share capital as registered with the Commercial Register on July 5, 2019, was increased by issuing 77 Class E preferred shares; |
| • | On August 22, 2019, our share capital as registered with the Commercial Register on August 28, 2019, was increased by an aggregate amount of CHF 3,714,300 through an increase of the par value of each of our Class A common shares and Class B, C, D and E preferred shares from CHF 100 to CHF 1,000; |
| • | On September 19, 2019, our share capital as registered with the Commercial Register on September 19, 2019, was increased by issuing 140 Class A common shares; |
| • | In the one-to-15,625 share split of all issued shares effected on September 19, 2019, each of our issued shares was split into 15,625 shares of the same class with a par value of CHF 0.064 per share; and |
| • | In the five-to-four reverse share split of all issued shares effected on April 24, 2020, each of our issued shares was consolidated into 0.8 shares of the same class with a par value of CHF 0.08 per share, and an aggregate of 44 common shares were converted into 6 Class C preferred shares, 12 Class D preferred shares and 26 Class E preferred shares, each with a par value of CHF 0.08. |
| • | conditional share capital (capital-actions conditionnel) for the purpose of issuing shares in connection with, among other things, (i) option and conversion rights granted in connection with warrants and convertible bonds of the Company or one of our subsidiaries or (ii) grants of rights to employees, members of our board of directors or consultants or to our subsidiaries or other persons providing services to the Company or a subsidiary to subscribe for new shares (conversion or option rights); or |
| • | authorized share capital (capital-actions autorisé) to be utilized by the board of directors within a period determined by the shareholders but not exceeding two years from the date of the shareholder approval. |
| • | if the issue price of the new registered shares is determined by reference to the market price; |
| • | for raising of capital (including private placements) in a fast and flexible manner, which would not be possible, or might only be possible with great difficulty or delays or at significantly less favorable conditions, without the exclusion of the statutory pre-emptive subscription rights of the existing shareholders; |
| • | for the acquisition of an enterprise, parts of an enterprise or participations, for the acquisition of products, intellectual property or licenses by or for investment projects of the Company or any of its group companies, or for the financing or refinancing of any of such transactions through a placement of shares; |
| • | for purposes of broadening the shareholder constituency of the Company in certain geographic, financial or investor markets, for purposes of the participation of strategic partners, or in connection with the listing of new shares on domestic or foreign stock exchanges; |
| • | for purposes of granting an over-allotment option or an option to purchase additional shares in a placement or sale of shares to the respective initial purchaser(s) or underwriter(s); |
| • | for the participation of members of the board of directors, members of the executive committee, employees, contractors, consultants or other persons performing services for the benefit of the Company or any of its group companies; |
| • | following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 20% of our share capital registered in the Commercial Register without having submitted to all other shareholders a takeover offer recommended by the board of directors; |
| • | for the defense of an actual, threatened or potential takeover bid, that the board of directors, upon consultation with an independent financial adviser retained by it, has not recommended to the shareholders acceptance on the basis that the board of directors has not found the takeover bid to be financially fair to the shareholders or not to be in the Company’s interest; or |
| • | for other valid grounds in the sense of Article 652b para. 2 of the CO. |
| • | for the purpose of financing or refinancing, or the payment for, the acquisition of enterprises, parts of enterprises, participations, intellectual property rights, licenses or investments; |
| • | if the issuance occurs in domestic or international capital markets, including private placements; |
| • | following a shareholder or a group of shareholders acting in concert having accumulated shareholdings in excess of 20% of the share capital registered in the Commercial Register without having submitted to all other shareholders a takeover offer recommended by the board of directors; or |
| • | for the defense of an actual, threatened or potential takeover bid that the board of directors, upon consultation with an independent financial adviser retained by it, has not recommended to the shareholders to accept on the basis that the board of directors has not found the takeover bid to be financially fair to the shareholders or not to be in the Company’s interest. |
| • | adopting and amending the articles of association, including the change of a company’s purpose or domicile; |
| • | electing the members of the board of directors, the chairman of the board of directors, the members of the compensation committee, the auditors and the independent proxy; |
| • | approving the business report, the annual statutory and consolidated financial statements, and deciding on the allocation of profits as shown on the balance sheet, in particular with regard to dividends; |
| • | approving the aggregate amount of compensation of members of the board of directors and the executive committee; |
| • | discharging the members of the board of directors and the executive committee from liability with respect to their conduct of business; |
| • | dissolving a company with or without liquidation; and |
| • | deciding matters reserved to the general meeting of shareholders by law or the articles of association or submitted to it by the board of directors. |
| • | amending the Company’s corporate purpose; |
| • | creating shares with preference rights; |
| • | cancelling or amending the transfer restrictions of shares; |
| • | creating authorized or conditional share capital; |
| • | increasing share capital out of equity, against contributions in-kind or for the purpose of acquiring specific assets and granting specific benefits; |
| • | limiting or withdrawing shareholder’s pre-emptive subscription rights; |
| • | changing a company’s domicile; |
| • | amending or repealing the voting and recording restrictions, the provision setting a maximum board size or the indemnification provision for the board of directors and the executive committee set forth in our articles of association; |
| • | converting registered shares into bearer shares; |
| • | removing the chairman or any member of the board of directors before the end of his or her term of office; and |
| • | dissolving or liquidating the Company. |
| • | a brief description of the business desired to be brought before the general meeting of shareholders and the reasons for conducting such business at the general meeting of shareholders; |
| • | the motions regarding the agenda item; |
| • | the name and address, as they appear in the share register, of the shareholder proposing such business; |
| • | the number of shares which are beneficially owned by such shareholder (including documentary support of such beneficial ownership); |
| • | the dates upon which the shareholder acquired such shares; |
| • | any material interest of the proposing shareholder in the proposed business; |
| • | a statement in support of the matter; and |
| • | all other information required under the applicable laws and stock exchange rules. |
| • | a core part of our business is sold without which it is economically impracticable or unreasonable to continue to operate the remaining business; |
| • | our assets, after the divestment, are not invested in accordance with our corporate purpose as set forth in the articles of association; and |
| • | the proceeds of the divestment are not earmarked for reinvestment in accordance with our corporate purpose but, instead, are intended for distribution to our shareholders or for financial investments unrelated to our corporate purpose. |
| • | the ultimate direction of the business of the Company and issuing of the relevant directives; |
| • | laying down the organization of the Company; |
| • | formulating accounting procedures, financial controls and financial planning; |
| • | nominating and removing persons entrusted with the management and representation of the Company and regulating the power to sign for the Company; |
| • | the ultimate supervision of those persons entrusted with management of the Company, with particular regard to adherence to law, our articles of association, and regulations and directives of the Company; |
| • | issuing the business report and the compensation report, and preparing for the general meeting of shareholders and carrying out its resolutions; and |
| • | informing the court in case of over-indebtedness. |
| • | severance payments (compensation due until the termination of a contractual relationship does not qualify as severance payment); |
| • | advance compensation; |
| • | incentive fees for the acquisition or transfer of companies, or parts thereof, by the Company or by companies being, directly or indirectly, controlled by us; |
| • | loans, other forms of indebtedness, pension benefits not based on occupational pension schemes and performance-based compensation not provided for in the articles of association; and |
| • | equity-based compensation not provided for in the articles of association. |
| • | the maximum aggregate amount of compensation of the board of directors for the term of office until the next annual general meeting of shareholders; and |
| • | the maximum aggregate amount of fixed compensation of the executive committee for the following financial year; and |
| • | the maximum aggregate amount of variable compensation of the executive committee for the current financial year. |
DELAWARE CORPORATE LAW | | | SWISS CORPORATE LAW |
Mergers and similar arrangements | |||
| | | ||
Under the Delaware General Corporation Law, with certain exceptions, a merger, consolidation, sale, lease or transfer of all or substantially all of the assets of a corporation must be approved by the board of directors and a majority of the outstanding shares entitled to vote thereon. A shareholder of a Delaware corporation participating in certain major corporate transactions may, under certain circumstances, be entitled to appraisal rights pursuant to which such shareholder may receive cash in the amount of the fair value of the shares held by such shareholder (as determined by a court) in lieu of the consideration such shareholder would otherwise receive in the transaction. The Delaware General Corporation Law also provides that a parent corporation, by resolution of its board of directors, may merge with any subsidiary, of which it owns at least 90.0% of each class of capital stock without a vote by the shareholders of such subsidiary. Upon any such merger, dissenting shareholders of the subsidiary would have appraisal rights. | | | Under Swiss law, with certain exceptions, a merger or a demerger of the corporation or a sale of all or substantially all of the assets of a corporation must be approved by two-thirds of the voting rights represented at the respective general meeting of shareholders as well as the absolute majority of the par value of shares represented at such general meeting of shareholders. A shareholder of a Swiss corporation participating in a statutory merger or demerger pursuant to the Swiss Merger Act (Loi sur la fusion) can file a lawsuit against the surviving company. If the consideration is deemed “inadequate,” such shareholder may, in addition to the consideration (be it in shares or in cash) receive an additional amount to ensure that such shareholder receives the fair value of the shares held by such shareholder. Swiss law also provides that if the merger agreement provides only for a compensation payment, at least 90.0% of all members in the transferring legal entity, who are entitled to vote, shall approve the merger agreement. |
| | | ||
Shareholders’ suits | |||
| | | ||
Class actions and derivative actions generally are available to shareholders of a Delaware corporation for, among other things, breach of fiduciary duty, corporate waste and actions not taken in accordance with applicable law. In such actions, the court has discretion to permit the winning party to recover attorneys’ fees incurred in connection with such action. | | | Class actions and derivative actions as such are not available under Swiss law. Nevertheless, certain actions may have a similar effect. A shareholder is entitled to bring suit against directors for breach of their duties and claim the payment of the company’s losses or damages to the corporation and, in some cases, to the individual shareholder. Likewise, an appraisal lawsuit won by a shareholder may indirectly compensate all shareholders. In addition, to the extent that U.S. laws and regulations provide a basis for liability and U.S. courts have jurisdiction, a class action may be available. |
| | | ||
| | | Under Swiss law, the winning party is generally entitled to recover a limited amount of attorneys’ fees incurred in connection with such action. The court has discretion to permit the shareholder who lost the lawsuit to recover attorneys’ fees incurred to the extent that he acted in good faith. | |
| | | ||
DELAWARE CORPORATE LAW | | | SWISS CORPORATE LAW |
Shareholder vote on board and management compensation | |||
| | | ||
Under the Delaware General Corporation Law, the board of directors has the authority to fix the compensation of directors, unless otherwise restricted by the certificate of incorporation or bylaws. | | | Pursuant to the Swiss Ordinance against excessive compensation in listed stock corporations (Ordonnance contre les rémunérations abusives dans les sociétés anonymes cotées en bourse), the general meeting of shareholders has the non-transferable right, amongst others, to vote separately and bindingly on the aggregate amount of compensation of the members of the board of directors, of the executive committee and of the advisory boards. |
| | | ||
Annual vote on board renewal | |||
| | | ||
Unless directors are elected by written consent in lieu of an annual meeting, directors are elected in an annual meeting of shareholders on a date and at a time designated by or in the manner provided in the bylaws. Re-election is possible. Classified boards are permitted. | | | The general meeting of shareholders elects the members of the board of directors, the chairperson of the board of directors and the members of the compensation committee individually and annually for a term of office until the end of the following general meeting of shareholders. Re-election is possible. |
| | | ||
Indemnification of directors and executive officers and limitation of liability | |||
| | | ||
The Delaware General Corporation Law provides that a certificate of incorporation may contain a provision eliminating or limiting the personal liability of directors (but not other controlling persons) of the corporation for monetary damages for breach of a fiduciary duty as a director, except no provision in the certificate of incorporation may eliminate or limit the liability of a director for: • any breach of a director’s duty of loyalty to the corporation or its shareholders; • acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law; • statutory liability for unlawful payment of dividends or unlawful share purchase or redemption; or • any transaction from which the director derived an improper personal benefit. A Delaware corporation may indemnify any person who was or is a party or is threatened to be made a party to any proceeding, other than an action by or on behalf of the corporation, because the person is or was a director or officer, against liability incurred in connection with the proceeding if the director or officer acted in good faith and in a manner reasonably | | | Under Swiss corporate law, an indemnification by the corporation of a director or member of the executive committee in relation to potential personal liability is not effective to the extent the director or member of the executive committee intentionally or negligently violated his or her corporate duties towards the corporation (certain views advocate that at least a grossly negligent violation is required to exclude the indemnification). Furthermore, the general meeting of shareholders may discharge (release) the directors and members of the executive committee from liability for their conduct to the extent the respective facts are known to shareholders. Such discharge is effective only with respect to claims of the company and of those shareholders who approved the discharge or who have since acquired their shares in full knowledge of the discharge. Most violations of corporate law are regarded as violations of duties towards the corporation rather than towards the shareholders. In addition, indemnification of other controlling persons is not permitted under Swiss corporate law, including shareholders of the corporation. The articles of association of a Swiss corporation may also set forth that the corporation shall indemnify and hold harmless, to the extent permitted by the law, the directors and executive managers out of assets of the corporation against threatened, pending or completed actions. |
DELAWARE CORPORATE LAW | | | SWISS CORPORATE LAW |
believed to be in, or not opposed to, the best interests of the corporation; and the director or officer, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful. Unless ordered by a court, any foregoing indemnification is subject to a determination that the director or officer has met the applicable standard of conduct: • by a majority vote of the directors who are not parties to the proceeding, even though less than a quorum; • by a committee of directors designated by a majority vote of the eligible directors, even though less than a quorum; • by independent legal counsel in a written opinion if there are no eligible directors, or if the eligible directors so direct; or • by the shareholders. Moreover, a Delaware corporation may not indemnify a director or officer in connection with any proceeding in which the director or officer has been adjudged to be liable to the corporation unless and only to the extent that the court determines that, despite the adjudication of liability but in view of all the circumstances of the case, the director or officer is fairly and reasonably entitled to indemnity for those expenses which the court deems proper. | | | Also, a corporation may enter into and pay for directors’ and officers’ liability insurance, which may cover negligent acts as well. |
| | | ||
Directors’ fiduciary duties | |||
| | | ||
A director of a Delaware corporation has a fiduciary duty to the corporation and its shareholders. This duty has two components: • the duty of care; and • the duty of loyalty. The duty of care requires that a director act in good faith, with the care that an ordinarily prudent person would exercise under similar circumstances. Under this duty, a director must inform himself or herself of, and disclose to shareholders, all material information reasonably available regarding a significant transaction. | | | The board of directors of a Swiss corporation manages the business of the corporation, unless responsibility for such management has been duly delegated to the executive committee based on organizational rules. However, there are several non-transferable duties of the board of directors: • the overall management of the corporation and the issuing of all necessary directives; • determination of the corporation’s organization; • the organization of the accounting, financial control and financial planning systems as required for management of the corporation; |
DELAWARE CORPORATE LAW | | | SWISS CORPORATE LAW | | ||
The duty of loyalty requires that a director act in a manner he or she reasonably believes to be in the best interests of the corporation. He or she must not use his or her corporate position for personal gain or advantage. This duty prohibits self-dealing by a director and mandates that the best interest of the corporation and its shareholders take precedence over any interest possessed by a director, officer or controlling shareholder and not shared by the shareholders generally. In general, actions of a director are presumed to have been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties. Should such evidence be presented concerning a transaction by a director, a director must prove the procedural fairness of the transaction, and that the transaction was of fair value to the corporation. | | | • the appointment and dismissal of persons entrusted with managing and representing the corporation; • overall supervision of the persons entrusted with managing the corporation, in particular with regard to compliance with the law, articles of association, operational regulations and directives; • compilation of the annual report, preparation for the general meeting of the shareholders, the compensation report and implementation of its resolutions; and • notification of the court in the event that the company is over-indebted. The members of the board of directors must perform their duties with all due diligence and safeguard the interests of the corporation in good faith. They must afford the shareholders equal treatment in equal circumstances. The duty of care requires that a director act in good faith, with the care that an ordinarily prudent director would exercise under like circumstances The duty of loyalty requires that a director safeguard the interests of the corporation and requires that directors act in the interest of the corporation and, if necessarily, put aside their own interests. If there is a risk of a conflict of interest, the board of directors must take appropriate measures to ensure that the interests of the company are duly taken into account. The burden of proof for a violation of these duties is with the corporation or with the shareholder bringing a suit against the director. | | ||
| | | | ||||
Shareholder action by written consent | | |||||
| | | | ||||
A Delaware corporation may, in its certificate of incorporation, eliminate the right of shareholders to act by written consent. | | | Shareholders of a Swiss corporation may only exercise their voting rights in a general meeting of shareholders and may not act by written consents. The articles of association must allow for (independent) proxies to be present at a general meeting of shareholders. The instruction of such (independent) proxies may occur in writing or electronically. | |||
| | | | ||||
Shareholder proposals | | |||||
| | | | ||||
A shareholder of a Delaware corporation has the right to put any proposal before the annual meeting of shareholders, provided it complies with the notice | | | At any general meeting of shareholders any shareholder may put proposals to the meeting if the proposal is part of an agenda item. No resolution may be taken on | | ||
DELAWARE CORPORATE LAW | | | SWISS CORPORATE LAW |
provisions in the governing documents. A special meeting may be called by the board of directors or any other person authorized to do so in the governing documents, but shareholders may be precluded from calling special meetings. | | | proposals relating to the agenda items that were not duly notified. Unless the articles of association provide for a lower threshold or for additional shareholders’ rights: • shareholders together representing at least 10% of the share capital may demand that a general meeting of shareholders be called for specific agenda items and specific proposals; and • shareholders together representing shares with a par value of at least CHF 1.0 million or 10% of the share capital, whichever is lower, may demand that an agenda item including a specific proposal be put on the agenda for a scheduled general meeting of shareholders, provided such request is made with appropriate lead time. Any shareholder can propose candidates for election as directors or make other proposals within the scope of an agenda item without prior written notice. In addition, any shareholder is entitled, at a general meeting of shareholders and without advance notice, to (i) request information from the board of directors on the affairs of the company (note, however, that the right to obtain such information is limited), (ii) request information from the auditors on the methods and results of their audit, (iii) request that the general meeting of shareholders resolve to convene an extraordinary general meeting, or (iv) request that the general meeting of shareholders resolve to appoint an examiner to carry out a special examination (“contrôle sp��cial”). |
| | | ||
Cumulative voting | |||
| | | ||
Under the Delaware General Corporation Law, cumulative voting for elections of directors is not permitted unless the corporation’s certificate of incorporation provides for it. | | | Cumulative voting is not permitted under Swiss corporate law. Pursuant to Swiss law, shareholders can vote for each proposed candidate, but they are not allowed to cumulate their votes for single candidates. An annual individual election of (i) all members of the board of directors, (ii) the chairperson of the board of directors, (iii) the members of the compensation committee, (iv) the election of the independent proxy for a term of office of one year (i.e., until the following annual general meeting of shareholders), as well as the vote on the aggregate amount of compensation of the members of the board of directors, of the executive committee and of the members of any advisory board, is mandatory for listed companies. Re-election is permitted. |
| | | ||
DELAWARE CORPORATE LAW | | | SWISS CORPORATE LAW |
Removal of directors | |||
| | | ||
A Delaware corporation with a classified board may be removed only for cause with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. | | | A Swiss corporation may remove, with or without cause, any director at any time with a resolution passed by a majority of the shares represented at a general meeting of shareholders. The articles of association may require the approval by a supermajority of the shares represented at a meeting for the removal of a director. |
| | | ||
Transactions with interested shareholders | |||
| | | ||
The Delaware General Corporation Law generally prohibits a Delaware corporation from engaging in certain business combinations with an “interested shareholder” for three years following the date that such person becomes an interested shareholder. An interested shareholder generally is a person or group who or which owns or owned 15.0% or more of the corporation’s outstanding voting shares within the past three years. | | | No such rule applies to a Swiss corporation. |
Dissolution; Winding up | |||
| | | ||
Unless the board of directors of a Delaware corporation approves the proposal to dissolve, dissolution must be approved by shareholders holding 100.0% of the total voting power of the corporation. Only if the dissolution is initiated by the board of directors may it be approved by a simple majority of the corporation’s outstanding shares. Delaware law allows a Delaware corporation to include in its certificate of incorporation a supermajority voting requirement in connection with dissolutions initiated by the board. | | | A dissolution of a Swiss corporation requires the approval by two-thirds of the voting rights represented at the respective general meeting of shareholders as well as the absolute majority of the par value of shares represented at such general meeting of shareholders. The articles of association may increase the voting thresholds required for such a resolution. |
Variation of rights of shares | |||
| | | ||
A Delaware corporation may vary the rights of a class of shares with the approval of a majority of the outstanding shares of such class, unless the certificate of incorporation provides otherwise. | | | The general meeting of shareholders of a Swiss corporation may resolve that preference shares be issued or that existing shares be converted into preference shares with a resolution passed by a majority of the shares represented at the general meeting of shareholders. Where a company has issued preference shares, further preference shares conferring preferential rights over the existing preference shares may be issued only with the consent of both a special meeting of the adversely affected holders of the existing preference shares and of a general meeting of all shareholders, unless otherwise provided in the articles of association. Shares with preferential voting rights are not regarded as preference shares for these purposes. |
| | | ||
DELAWARE CORPORATE LAW | | | SWISS CORPORATE LAW |
Amendment of governing documents | |||
| | | ||
A Delaware corporation’s governing documents may be amended with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. | | | The articles of association of a Swiss corporation may be amended with a resolution passed by a majority of the shares represented at a general meeting of shareholders, unless otherwise provided in the articles of association. There are a number of resolutions, such as an amendment of the stated purpose of the corporation, the introduction of authorized and conditional capital and the introduction of shares with preferential voting rights that require the approval by two-thirds of the votes and an absolute majority of the par value of the shares represented at such general meeting of shareholders. The articles of association may increase these voting thresholds. |
| | | ||
Inspection of books and records | |||
| | | ||
Shareholders of a Delaware corporation, upon written demand under oath stating the purpose thereof, have the right during the usual hours for business to inspect for any proper purpose, and to obtain copies of list(s) of shareholders and other books and records of the corporation and its subsidiaries, if any, to the extent the books and records of such subsidiaries are available to the corporation. | | | Shareholders of a Swiss corporation may only inspect books and records if the general meeting of shareholders or the board of directors approved such inspection. The information may be refused where providing it would jeopardize the corporation’s trade secrets or other interests warranting protection. A shareholder is only entitled to receive information to the extent required to exercise his or her rights as a shareholder, subject to the interests of the corporation. A shareholder’s right to inspect the share register is limited to the right to inspect his or her own entry in the share register. |
| | | ||
Payment of dividends | |||
| | | ||
The board of directors may approve a dividend without shareholder approval. Subject to any restrictions contained in its certificate of incorporation, the board may declare and pay dividends upon the shares of its capital stock either: • out of its surplus, or • in case there is no such surplus, out of its net profits for the fiscal year in which the dividend is declared and/or the preceding fiscal year. Shareholder approval is required to authorize capital stock in excess of that provided in the charter. Directors may issue authorized shares without shareholder approval. | | | Dividend payments are subject to the approval of the general meeting of shareholders. The board of directors may propose to shareholders that a dividend shall be paid but cannot itself authorize the distribution. Payments out of a corporation’s share capital (in other words, the aggregate par value of the corporation’s registered share capital) in the form of dividends are not allowed and may be made only by way of a share capital reduction. Dividends may be paid only from the profits of the previous business year or brought forward from previous business years or if the corporation has distributable reserves, each as evidenced by the corporation’s audited stand-alone statutory balance sheet prepared pursuant to Swiss law and after allocations to reserves required by Swiss law and the articles of association have been deducted. |
| | | ||
DELAWARE CORPORATE LAW | | | SWISS CORPORATE LAW |
Creation and issuance of new shares | |||
| | | ||
All creation of shares require the board of directors to adopt a resolution or resolutions, pursuant to authority expressly vested in the board of directors by the provisions of the company’s certificate of incorporation. | | | All creation of shares require a shareholders’ resolution. The creation of authorized or contingent share capital requires at least two-thirds of the voting rights represented at the general meeting of shareholders and an absolute majority of the par value of shares represented at such meeting. The board of directors may issue shares out of the authorized share capital during a period of up to two years. Shares are created and issued out of contingent share capital through the exercise of options or of conversion rights that the board of director may grant in relation to, e.g., debt instruments or employees. |
| • | 1% of the number of our common shares then outstanding, which will equal approximately 639,322 common shares immediately after this offering, assuming no exercise of the underwriters’ option to purchase additional common shares; or |
| • | the average weekly trading volume of our common shares on the NYSE during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale; |
| • | certain banks, insurance companies and other financial institutions; |
| • | brokers, dealers or traders in securities who use a mark-to-market method of tax accounting; |
| • | persons holding common shares as part of a hedging transaction, straddle, wash sale, conversion transaction or other integrated transaction or persons entering into a constructive sale with respect to the common shares; |
| • | persons whose functional currency for U.S. federal income tax purposes is not the U.S. dollar; |
| • | entities or arrangements classified as partnerships or S corporations for U.S. federal income tax purposes; |
| • | tax-exempt entities, including an “individual retirement account” or “Roth IRA” or governmental entities; |
| • | real estate investment trusts or regulated investment companies; |
| • | former U.S. citizens or long-term residents of the United States; |
| • | persons subject to Section 451(b) of the Code; |
| • | persons that own or are deemed to own 10% or more of the voting power or value of our shares; or |
| • | persons holding common shares in connection with a trade or business conducted outside of the United States or in connection with a permanent establishment or other fixed place of business outside of the United States. |
| • | a citizen or individual resident of the United States; |
| • | a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or the District of Columbia; or |
| • | an estate or trust the income of which is subject to U.S. federal income taxation regardless of its source. |
Underwriter | | | Number of Common Shares |
Morgan Stanley & Co. LLC | | | |
BofA Securities, Inc. | | | |
Cowen and Company, LLC | | | |
Total | | | 7,355,000 |
| | | | | Total | |||||
| | | Per Share | | | No Exercise | | | Full Exercise | |
Public offering price | | | $ | | | $ | | | $ |
Underwriting discounts and commissions | | | $ | | | $ | | | $ |
Proceeds, before expenses, to us | | | $ | | | $ | | | $ |
| • | offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, lend or otherwise transfer or dispose of, directly or indirectly, any common shares or any securities convertible into or exercisable or exchangeable for common shares; |
| • | file any registration statement with the SEC relating to the offering of any common shares or any securities convertible into or exercisable or exchangeable for common shares; or |
| • | enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of the common shares, |
| (i) | transactions relating to common shares or any security convertible into common shares acquired in the offering (other than any issuer-directed common shares purchased in the offering by our officers or directors) or in open market transactions after the completion of the offering; |
| (ii) | transfers or distributions as a bona fide gift or for bona fide estate planning purposes or to a charitable organization or educational institution; |
| (iii) | transfers or distributions to any immediate family member of such person, affiliate or any trust or trustee or beneficiary thereof for the direct or indirect benefit of such person or the immediate family of such person (for this purpose, “immediate family” means any relationship by blood, marriage, domestic partnership or adoption, not more remote than first cousin); |
| (iv) | transfers or distributions to any corporation, partnership, limited liability company or other entity or affiliate of such person or the immediate family of such person; |
| (v) | transfers or distributions (a) by will, other testamentary document or intestate succession to the legal representative, heir, beneficiary or a member of the immediate family of such person upon the death of such person; (b) by operation of law pursuant to a domestic order or negotiated divorce settlement; |
| (vi) | transfers or distributions to another corporation, member, partnership, limited liability company, trust or other entity that is a direct or indirect affiliate (as defined under Rule 12b-2 of the Exchange Act), or to an investment fund or other entity that controls or manages, or is under common control with, such person, or distributions to partners, members, shareholders, beneficiaries or other equity holders of such person; |
| (vii) | transfers or distributions to us (a) in connection with the repurchase of such securities with respect to the termination of such person’s employment with us or (b) pursuant to contractual arrangements described in this prospectus; |
| (viii) | transfers or distributions to us in connection with the repayment (including any adjustments) of any loans or promissory notes of such as described in this prospectus; |
| (ix) | transfers or distributions (including through a “cashless” exercise or on a “net exercise basis”) to us in connection with the conversion of any convertible security into, or the exercise of any option or warrant for, common shares (including to satisfy withholding obligations or the payment of taxes in connection therewith); provided that (a) any such common shares received by such person shall be subject to the lock-up agreement and (b) no filing under Section 16(a) of the Exchange Act (or its foreign equivalent) reporting a reduction in beneficial ownership of common shares shall be required or shall be voluntarily made during the restricted period; |
| (x) | transfers or distributions prior to the date of the public filing of the registration statement of which this prospectus forms a part and pursuant to our shareholders’ agreement; |
| (xi) | transfers or distributions to a nominee or custodian of a person or entity to whom a disposition or transfer would be permissible under clauses (i) through (x) above, provided that any common shares shall be subject to the terms of the lock-up agreement; |
| (xii) | any pledge or transfer by such person (or any permitted transferee) of common shares or any security convertible into common shares pursuant to agreements governing indebtedness or commitments relating to indebtedness of such person (or any permitted transferee) or its affiliates (other than us and our subsidiaries) in effect on the date thereof (and any refinancing or replacement thereof) and described in this prospectus and any transfer upon foreclosure, provided that any required filing under Section 16(a) of the Exchange Act (or its foreign equivalent) reporting a reduction in beneficial ownership by such person or any party (pledgor or pledgee) shall indicate by footnote disclosure or otherwise the nature of the transfer and if any filing is required to be made under Section 16(a) of the Exchange Act (or its foreign equivalent) during the restricted period, and such person shall provide the representatives prior written notice informing them of such report; |
| (xiii) | the establishment of a trading plan pursuant to Rule 10b5-1 under the Exchange Act (or its foreign equivalent) for the transfer of common shares, provided that (a) such plan does not provide for the transfer of common shares during the restricted period and (b) to the extent a public announcement or filing under the Exchange Act (or its foreign equivalent), if any, is required of or voluntarily made by or on behalf of such person or us regarding the establishment of such plan, such announcement or filing shall include a statement to the effect that no transfer of common shares may be made under such plan during the restricted period; |
| (xiv) | transfers or dispositions pursuant to a bona fide tender offer for our capital shares, merger, consolidation or other similar transaction made to all holders of our securities involving a change of control of us (including without limitation, the entering into of any lock-up, voting or similar agreement pursuant to which such person may agree to transfer, sell, tender or otherwise dispose of common shares or any security convertible into common shares in connection with such transaction) that has been approved by our board of directors; provided that, in the event that such change of control transaction is not consummated, this paragraph shall not be applicable and such person’s shares and other securities shall remain subject to the lock-up agreement (for this purpose, “change of control” means the transfer whether by tender offer, merger, consolidation or other similar transaction), in one transactions or a series of related transactions, to a person or group of affiliated persons (other than the underwriters pursuant to this offering), of our voting securities if, after such transfer, such person or group of affiliated persons would hold greater than 50% of our outstanding voting securities); or |
| (xv) | the conversion, exercise or exchange of our preferred shares, options to purchase common shares, warrants or any security convertible into common shares pursuant to any reorganization, conversion or share split, as such terms are described in this prospectus; provided that any such securities shall remain subject to the lock-up agreement, |
| (i) | the common shares to be sold in this offering; |
| (ii) | the issuance by us of shares of common shares upon the exercise of an option or warrant or the conversion of a security outstanding on the date of this prospectus of which the underwriters have been advised in writing; |
| (iii) | the grant of any options to purchase common shares, restricted shares or restricted units under an incentive compensation plan in effect or approved by our board of directors on the date of this prospectus and described in this prospectus; |
| (iv) | our filing of any registration statement on Form S-8 or a successor form relating to the common shares granted pursuant to or reserved for issuance under an incentive compensation plan described in this prospectus; |
| (v) | offers to acquire, transfers, issuances or repurchases pursuant to loan settlement agreements, including the transfer of common shares to us in connection therewith, as described in this prospectus; |
| (vi) | the offer or issuance of common shares in connection with an acquisition, joint venture, commercial or collaborative relationship, or an acquisition or license by us of assets of another person or entity or pursuant to an employee benefit plan assumed by us in connection with any such acquisition, provided that (1) the aggregate number of shares issued does not exceed 5% of the total number of outstanding shares of our common shares immediately following the closing of this offering and (2) the recipient of any such shares during the restricted period enters into a lock-up agreement; |
| (vii) | facilitating the establishment of a trading plan on behalf of one of our shareholders, officers or directors pursuant to Rule 10b5-1 under the Exchange Act for the transfer of common shares, provided that such plan does not provide for the transfer of common shares during the restricted period and, to the extent a public announcement or filing under the Exchange Act is required of or voluntarily made by us regarding the establishment of such plan, such announcement or filing shall include a statement to the effect that no transfer of common shares may be made under such plan during the restricted period; |
| (viii) | the issuance and sale of securities convertible into common shares pursuant to the Facility Agreement and the issuance of common shares upon the conversion of such securities; or |
| (ix) | the confidential submission of a registration statement with the SEC with respect to common shares issued and issuable upon the conversion of securities described in clause (viii) above exclusively as a result of contractual obligations contained in the Registration Rights Agreement to be entered into in connection with the Facility Agreement. |
| (a) | to any legal entity which is a qualified investor as defined in the Prospectus Regulation; |
| (b) | to fewer than 150 natural or legal persons (other than qualified investors as defined in the Prospectus Regulation), subject to obtaining the prior consent of the representatives for any such offer; or |
| (c) | in any other circumstances falling within Article 1(4) of the Prospectus Regulation, |
| (a) | a corporation (which is not an accredited investor (as defined in Section 4A of the SFA)) the sole business of which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or |
| (b) | a trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary of the trust is an individual who is an accredited investor, |
| (a) | to an institutional investor or to a relevant person defined in Section 275(2) of the SFA, or to any person arising from an offer referred to in Section 275(1A) or Section 276(4)(i)(B) of the SFA; |
| (b) | where no consideration is or will be given for the transfer; |
| (c) | where the transfer is by operation of law; |
| (d) | as specified in Section 276(7) of the SFA; or |
| (e) | as specified in Regulation 37A of the Securities and Futures (Offers of Investments) (Securities and Securities-based Derivatives Contracts) Regulations 2018 of Singapore. |
| (a) | it has only communicated or caused to be communicated and will only communicate or cause to be communicated an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the Financial Services and Markets Act 2000 (“FSMA”) received by it in connection with the issue or sale of our common shares in circumstances in which Section 21(1) of the FSMA does not apply to us; and |
| (b) | it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to our common shares in, from or otherwise involving the United Kingdom. |
Expenses | | | Amount |
SEC registration fee | | | $19,762 |
NYSE listing fees and expenses | | | 200,000 |
FINRA filing fee | | | 23,338 |
Printing and engraving expenses | | | 150,000 |
Legal fees and expenses | | | 1,750,000 |
Accounting fees and expenses | | | 500,000 |
Stamp duty | | | 1,250,000 |
Miscellaneous costs | | | 1,106,900 |
Total | | | $5,000,000 |
| * | To be provided by amendment. |
| • | the non-Swiss court had jurisdiction pursuant to the PILA; |
| • | the judgment of such non-Swiss court has become final and non-appealable; |
| • | the judgment does not contravene Swiss public policy; |
| • | the court procedures and the service of documents leading to the judgment were in accordance with the due process of law; and |
| • | no proceeding involving the same position and the same subject matter was first brought in Switzerland, or adjudicated in Switzerland, or was earlier adjudicated in a third state and this decision is recognizable in Switzerland. |
| | | ||
| | | ||
| | | ||
| | | ||
Consolidated Financial Statements | | | |
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | |
| | | Note | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 | |
Contract revenue | | | 7 | | | 2,340 | | | 1,140 |
Research and development expenses | | | 9/11 | | | (107,537) | | | (118,313) |
General and administrative expenses | | | 11 | | | (14,202) | | | (8,768) |
Operating loss | | | | | (119,399) | | | (125,941) | |
Other income | | | 8 | | | 1,655 | | | — |
Financial income | | | | | 2,253 | | | 2,856 | |
Financial expense | | | | | (156) | | | — | |
Exchange differences | | | | | (255) | | | 213 | |
Loss before taxes | | | | | (115,902) | | | (122,872) | |
Income tax expenses | | | 12 | | | (582) | | | (224) |
Loss for the year | | | | | (116,484) | | | (123,096) | |
| | | | | | | ||||
Attributable to: | | | | | | | |||
Owners of the parent | | | | | (116,484) | | | (123,096) | |
| | | | | | | ||||
Loss per share | | | | | | | |||
Basic and diluted loss per share (in USD) | | | 28 | | | (2.36) | | | (2.64) |
| | | Note | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 | |
Loss for the year | | | | | (116,484) | | | (123,096) | |
| | | | | | |||||
Other comprehensive (loss) / profit: | | | | | | | |||
Items that will not be reclassified to profit or loss | | | | | | | |||
Remeasurements of defined benefit plan | | | 22 | | | (1,346) | | | (193) |
Total items that will not be reclassified to profit or loss | | | | | (1,346) | | | (193) | |
Items that may be reclassified subsequently to profit or loss | | | | | | | |||
Currency translation differences | | | | | 112 | | | (79) | |
Total items that may be reclassified subsequently to profit or loss | | | | | 112 | | | (79) | |
| | | | | | | ||||
Other comprehensive loss for the year | | | | | (1,234) | | | (272) | |
| | | | | | | ||||
Total comprehensive loss for the year | | | | | (117,718) | | | (123,368) | |
| | | | | | | ||||
Attributable to: | | | | | | | |||
Owners of the parent | | | | | (117,718) | | | (123,368) |
| | | Note | | | December 31, 2019 | | | December 31, 2018 | |
ASSETS | | | | | | | |||
Current assets | | | | | | | |||
Cash and cash equivalents | | | 19 | | | 115,551 | | | 138,807 |
Trade accounts receivable | | | 13/19/27 | | | — | | | 192 |
Other current assets | | | 14 | | | 7,055 | | | 3,081 |
Total current assets | | | | | 122,606 | | | 142,080 | |
Non-current assets | | | | | | | |||
Property, plant and equipment | | | 16 | | | 1,376 | | | 1,540 |
Right-of-use assets | | | 4/17 | | | 4,898 | | | — |
Intangible assets | | | 18 | | | 8,434 | | | 6,674 |
Other long-term assets | | | | | 368 | | | 264 | |
Total non-current assets | | | | | 15,076 | | | 8,478 | |
| | | | | | | ||||
Total assets | | | | | 137,682 | | | 150,558 | |
| | | | | | | ||||
LIABILITIES AND EQUITY | | | | | | | |||
Current liabilities | | | | | | | |||
Trade accounts payable | | | 19a/27 | | | 3,329 | | | 6,750 |
Accrued liabilities and other payables | | | 21 | | | 15,430 | | | 13,650 |
Contract liability (short-term) | | | 7 | | | — | | | 1,765 |
Lease liabilities (short-term) | | | 4/17 | | | 1,132 | | | — |
Current income tax payable | | | | | 52 | | | 189 | |
Total current liabilities | | | | | 19,943 | | | 22,354 | |
Non-current liabilities | | | | | | | |||
Contract liability (long-term) | | | 7 | | | — | | | 575 |
Lease liabilities (long-term) | | | 4/17 | | | 3,899 | | | — |
Defined benefit pension liabilities | | | 22 | | | 2,684 | | | 1,386 |
Total non-current liabilities | | | | | 6,583 | | | 1,961 | |
| | | | | | | ||||
Total liabilities | | | | | 26,526 | | | 24,315 | |
| | | | | | | ||||
Equity attributable to owners of the parent | | | | | | | |||
Share capital | | | 24 | | | 4,361 | | | 401 |
Share premium | | | 24 | | | 549,922 | | | 452,268 |
Treasury shares | | | 24 | | | (100) | | | — |
Other reserves | | | 22/23 | | | 5,473 | | | 5,702 |
Cumulative translation adjustments | | | | | 69 | | | (43) | |
Accumulated losses | | | | | (448,569) | | | (332,085) | |
Total equity | | | | | 111,156 | | | 126,243 | |
| | | | | | | ||||
Total liabilities and equity | | | | | 137,682 | | | 150,558 |
| | | | | Attributable to owners of the parent | ||||||||||||||||||||
| | | Note | | | Share capital | | | Share premium | | | Other reserves | | | Treasury shares | | | Cumulative translation adjustment | | | Accumulated losses | | | Total | |
January 1, 2018 | | | | | 397 | | | 452,296 | | | 5,426 | | | — | | | 36 | | | (208,989) | | | 249,166 | |
Loss for the year | | | | | — | | | — | | | — | | | — | | | — | | | (123,096) | | | (123,096) | |
Translation adjustment | | | | | — | | | — | | | — | | | — | | | (79) | | | — | | | (79) | |
Remeasurements of defined benefit pension | | | 22 | | | — | | | — | | | (193) | | | — | | | — | | | — | | | (193) |
Total other comprehensive loss | | | | | — | | | — | | | (193) | | | — | | | (79) | | | — | | | (272) | |
Total comprehensive loss for the year | | | | | — | | | — | | | (193) | | | — | | | (79) | | | (123,096) | | | (123,368) | |
Issue of share capital | | | 24 | | | 4 | | | — | | | — | | | — | | | — | | | — | | | 4 |
Transaction costs | | | 24 | | | — | | | (28) | | | — | | | — | | | — | | | — | | | (28) |
Share-based compensation expense | | | 23 | | | — | | | — | | | 469 | | | — | | | — | | | — | | | 469 |
Total transactions with owners | | | | | 4 | | | (28) | | | 469 | | | — | | | — | | | — | | | 445 | |
December 31, 2018 | | | | | 401 | | | 452,268 | | | 5,702 | | | — | | | (43) | | | (332,085) | | | 126,243 | |
Loss for the year | | | | | — | | | — | | | — | | | — | | | — | | | (116,484) | | | (116,484) | |
Translation adjustment | | | | | — | | | — | | | — | | | — | | | 112 | | | — | | | 112 | |
Remeasurements of defined benefit pension | | | 22 | | | — | | | — | | | (1,346) | | | — | | | — | | | — | | | (1,346) |
Total other comprehensive loss | | | | | — | | | — | | | (1,346) | | | — | | | 112 | | | — | | | (1,234) | |
Total comprehensive loss for the year | | | | | — | | | — | | | (1,346) | | | — | | | 112 | | | (116,484) | | | (117,718) | |
Issue of share capital / capital contributions | | | 24 | | | 171 | | | 103,221 | | | — | | | — | | | — | | | — | | | 103,392 |
Transaction costs | | | 24 | | | — | | | (1,778) | | | — | | | — | | | — | | | — | | | (1,778) |
Transfer from share premium for par value increase | | | 24 | | | 3,789 | | | (3,789) | | | — | | | — | | | — | | | — | | | — |
Purchase of treasury shares | | | 24 | | | — | | | — | | | — | | | (141) | | | — | | | — | | | (141) |
Sale of treasury shares | | | 24 | | | — | | | — | | | — | | | 41 | | | — | | | — | | | 41 |
Share-based compensation expense | | | 23 | | | — | | | — | | | 1,117 | | | — | | | — | | | — | | | 1,117 |
Total transactions with owners | | | | | 3,960 | | | 97,654 | | | 1,117 | | | (100) | | | — | | | — | | | 102,631 | |
December 31, 2019 | | | | | 4,361 | | | 549,922 | | | 5,473 | | | (100) | | | 69 | | | (448,569) | | | 111,156 | |
| | | Note | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 | |
Cash used in operating activities | | | | | | | |||
Loss for the year | | | | | (116,484) | | | (123,096) | |
Adjustments for non-monetary items: | | | | | | | |||
Share-based compensation expense | | | 23 | | | 1,117 | | | 469 |
Depreciation of property, plant and equipment | | | 16 | | | 552 | | | 488 |
Depreciation of right-of-use assets | | | 17 | | | 1,064 | | | — |
Amortization and impairment of intangible assets | | | 18 | | | 30 | | | 252 |
Change in defined benefit pension liabilities | | | 22 | | | (53) | | | 119 |
Change in fair value measurement | | | | | 557 | | | 1,009 | |
Accrued withholding tax | | | | | 626 | | | 711 | |
Accrued R&D credit | | | | | (437) | | | — | |
Financial income | | | | | (2,253) | | | (2,856) | |
Financial expense (including interest on lease obligations) | | | | | 156 | | | — | |
Exchange differences | | | | | 128 | | | (29) | |
Income taxes | | | 12 | | | 582 | | | 224 |
Operating loss before working capital changes | | | | | (114,415) | | | (122,709) | |
Decrease in trade accounts receivable | | | | | 192 | | | 895 | |
(Increase) in other current assets | | | | | (4,030) | | | (1,678) | |
(Decrease) in contract liability (short and long term) | | | | | (2,340) | | | (1,140) | |
(Decrease) in trade accounts payable | | | | | (3,425) | | | (858) | |
Increase in accrued liabilities and other payables | | | | | 1,720 | | | 3,262 | |
Cash used in operating activities | | | | | (122,298) | | | (122,228) | |
| | | | | | | ||||
Interest received | | | | | 1,164 | | | 1,051 | |
Interest paid | | | | | (157) | | | — | |
Tax paid | | | | | (290) | | | (185) | |
Net cash used in operating activities | | | | | (121,581) | | | (121,362) | |
| | | | | | | ||||
Cash used in investing activities | | | | | | | |||
Payment for purchases of property, plant and equipment | | | 16 | | | (358) | | | (944) |
Payment for purchases of intangible assets | | | 18 | | | (1,790) | | | (1,526) |
Payment for rent deposits | | | | | (100) | | | (36) | |
Net cash used in investing activities | | | | | (2,248) | | | (2,506) | |
| | | | | | | ||||
Cash from / (used in) financing activities | | | | | | | |||
Proceeds from capital contributions, net of transaction costs | | | 24 | | | 101,614 | | | (24) |
Acquisition of treasury shares | | | 24 | | | (141) | | | — |
Sale of treasury shares | | | 24 | | | 41 | | | — |
Principal portion of lease obligations payments | | | 17 | | | (1,002) | | | — |
Net cash from / (used in) financing activities | | ��� | | | 100,512 | | | (24) | |
| | | | | | | ||||
Net decrease in cash and cash equivalents | | | | | (23,317) | | | (123,892) | |
Exchange gains / (losses) on cash and cash equivalents | | | | | 61 | | | (53) | |
Cash and cash equivalents at beginning of year | | | | | 138,807 | | | 262,752 | |
Cash and cash equivalents at end of year | | | | | 115,551 | | | 138,807 |
| Corporate information |
| 2. | Basis of preparation |
| - | establish and maintain a strong patent position and protection; |
| - | enter into collaborations with partners in the pharmaceutical industry; |
| - | acquire and retain key personnel; and |
| - | acquire additional funding to support its operations. |
| 3. | Significant accounting policies |
| 3.1 | Consolidation |
| 3.2 | Foreign currency translation |
| (i) | assets and liabilities for each balance sheet presented are translated at the closing rate at the date of that balance sheet; |
| (ii) | income and expenses for each income statement are translated at monthly average exchange rates; and |
| (iii) | all resulting exchange differences are recognized in other comprehensive income, under “Cumulative translation adjustments”. |
| 3.3 | Cash and cash equivalents |
| 3.4 | Property, plant and equipment |
Leasehold improvements | | | 3 to 10 years |
Laboratory equipment | | | 5 years |
Office equipment | | | 5 years |
Hardware | | | 3 years |
| 3.5 | Intangible assets |
| 3.6 | Impairment of non-financial assets |
| 3.7 | Employee benefits |
| - | including any market performance conditions; |
| - | excluding the impact of any service and non-market performance vesting conditions; and |
| - | including the impact of any non-vesting conditions. |
| 3.8 | Share capital and share premium |
| 3.9 | Treasury shares |
| 3.10 | Leases |
| - | the amount of the initial measurement of lease liability; |
| - | any lease payments made at or before the commencement date less any lease incentives received; |
| - | any initial direct costs, and |
| - | restoration costs. |
| - | periods covered by an option to extend the lease if the Company is reasonably certain to exercise that option; and |
| - | periods covered by an option to terminate the lease if the Company is reasonably certain not to exercise that option. |
| 3.11 | Revenue recognition |
| 3.12 | Research and development expenses |
| 3.13 | Current, deferred income tax and tax credit |
| 3.14 | Segment reporting |
| 3.15 | Loss per share |
| 4. | New and amended IFRS standards |
(in KUSD) | | | |
Operating lease commitments at December 31, 2018 | | | 4,378 |
| | | ||
Discounted at the incremental borrowing rate as at January 1, 2019 | | | 3,976 |
Short-term leases recognized on a straight-line basis as expenses | | | (15) |
Low-value leases recognized on a straight-line basis as expenses | | | — |
Extension options reasonably certain to be exercised | | | 1,462 |
Lease obligations recognized at January 1, 2019 | | | 5,423 |
| | | ||
Of which are: | | | |
Lease liabilities (short-term) | | | 924 |
Lease liabilities (long-term) | | | 4,499 |
(in KUSD) | | | December 31, 2019 | | | January 1, 2019 |
Properties (offices) | | | 4,820 | | | 5,399 |
Vehicles | | | 78 | | | 24 |
Total right-of-use assets | | | 4,898 | | | 5,423 |
| 5. | Financial risk management |
| 5.1 | Financial risk factors |
| - | forecast costs denominated in a currency other than the entity’s functional currency; |
| - | recognized assets and liabilities denominated in a currency other than the entity's functional currency; and |
| - | net investments in foreign operations. |
December 31 | | | 2019 in KL/C(1) | | | 2019 in KUSD | | | 2018 in KL/C(1) | | | 2018 in KUSD |
In USD | | | 109,939 | | | 109,939 | | | 135,451 | | | 135,451 |
In CHF | | | 457 | | | 472 | | | 1,143 | | | 1,160 |
In GBP | | | 3,529 | | | 4,654 | | | 929 | | | 1,179 |
In EUR | | | 433 | | | 486 | | | 889 | | | 1,017 |
| | | | | 115,551 | | | | | 138,807 |
| (1) | Thousands Local Currencies |
(in KUSD) | | | Note | | | Less than 3 months | | | Between 3 months and 1 year | | | Between 1 year and 3 years | | | More than 3 years |
Trade accounts payable | | | | | 3,266 | | | 63 | | | — | | | — | |
Lease liabilities | | | 17 | | | 311 | | | 935 | | | 1,859 | | | 2,265 |
At December 31, 2019 | | | | | 3,577 | | | 998 | | | 1,859 | | | 2,265 | |
| | | | | | | | | | | ||||||
Trade accounts payable | | | | | 6,597 | | | 153 | | | — | | | — | |
Lease liabilities | | | 17 | | | — | | | — | | | — | | | — |
At December 31, 2018 | | | | | 6,597 | | | 153 | | | — | | | — |
| 5.2 | Capital management |
| 5.3 | Fair value estimation |
| - | Cash and cash equivalents |
| - | Trade accounts receivable |
| - | Trade accounts payable |
| 6 | Critical accounting estimates and judgements |
| 7. | Contract revenue and contract liability |
(in KUSD) | | | 2019 | | | 2018 |
January 1 | | | 2,340 | | | 3,480 |
Recognized as contract revenue | | | (2,340) | | | (1,140) |
December 31 | | | — | | | 2,340 |
| | | | | |||
of which: | | | | | ||
Contract liability (short term) | | | — | | | 1,765 |
Contract liability (long term) | | | — | | | 575 |
Contract liability (total) | | | — | | | 2,340 |
| 8. | Other Income |
(in KUSD): | | | | | ||
Years | | | December 31, 2019 | | | December 31, 2018 |
2016 | | | 296 | | | — |
2017 | | | 350 | | | — |
2018 | | | 477 | | | — |
2019 | | | 532 | | | — |
| | | 1,655 | | | — |
| 9. | Research and development expenses |
| 10 | Employee expenses |
(in KUSD) | | | Note | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 |
| | | | | | | ||||
Wages, salaries and other costs | | | | | 24,061 | | | 19,245 | |
Social security costs | | | | | 3,871 | | | 3,452 | |
Share-based compensation expense | | | 23 | | | 1,117 | | | 469 |
Defined benefit plan - pension costs | | | 22 | | | 462 | | | 535 |
Defined contribution plan - pension costs | | | | | 540 | | | 481 | |
Employee expenses | | | | | 30,051 | | | 24,182 |
| 11 | Expenses by nature |
(in KUSD) | | | Note | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 |
R&D | | | | | | | |||
External costs | | | | | 81,363 | | | 98,493 | |
Employee expenses | | | 10 | | | 24,916 | | | 19,246 |
Depreciation of property, plant and equipment | | | 16 | | | 407 | | | 330 |
Depreciation of right-of-use assets | | | 17 | | | 837 | | | — |
Amortization of intangible assets | | | 18 | | | 14 | | | 17 |
Impairment of intangible assets | | | 18 | | | — | | | 227 |
Research and development expenses | | | | | 107,537 | | | 118,313 | |
| | | | | | | ||||
G&A | | | | | | | |||
External costs | | | | | 8,668 | | | 3,628 | |
Employee expenses | | | 10 | | | 5,135 | | | 4,936 |
General and administrative costs charged by related parties | | | 27 | | | 11 | | | 38 |
Depreciation of property, plant and equipment | | | 16 | | | 145 | | | 158 |
Depreciation of right-of-use assets | | | 17 | | | 227 | | | — |
Amortization of intangible assets | | | 18 | | | 16 | | | 8 |
General and administrative expenses | | | | | 14,202 | | | 8,768 | |
Total expenses by nature | | | | | 121,739 | | | 127,081 |
| 12 | Income tax expenses |
(in KUSD) | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 |
Current income taxes for the year | | | 572 | | | 218 |
Current income taxes related to prior years | | | 10 | | | 6 |
Income tax expenses | | | 582 | | | 224 |
(in KUSD) | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 |
Loss before taxes | | | 115,902 | | | 122,872 |
Tax calculated at tax domestic rates applicable to profits in the respective countries | | | (12,332) | | | (13,321) |
| | | | | |||
Tax effects of: | | | | | ||
- Tax losses for which no deferred income tax asset was recognized | | | 13,187 | | | 13,766 |
- Utilization of R&D tax credit (USA) | | | (436) | | | (310) |
- Income not subject to tax / (expenses not deductible for tax purposes) | | | 156 | | | 83 |
- Tax relating to prior years | | | 10 | | | 6 |
- Other | | | (3) | | | — |
Income tax expenses | | | 582 | | | 224 |
| 13. | Trade accounts receivable |
(in KUSD) | | | December 31, 2019 | | | December 31, 2018 |
Trade accounts receivable | | | — | | | 192 |
Less: Allowance for doubtful accounts | | | — | | | — |
Trade accounts receivable, net | | | — | | | 192 |
| 14. | Other current assets |
(in KUSD) | | | December 31, 2019 | | | December 31, 2018 |
VAT receivable, net | | | 471 | | | 549 |
Withholding tax receivable | | | 626 | | | 968 |
Prepaid expenses | | | 4,215 | | | 1,384 |
UK R&D expenditure credits | | | 891 | | | — |
Other | | | 852 | | | 180 |
| | | 7,055 | | | 3,081 |
| 15. | Non-current assets by geographic area |
(in KUSD): | | | | | ||
Country | | | December 31, 2019 | | | December 31, 2018 |
Switzerland | | | 10,903 | | | 7,112 |
United Kingdom | | | 2,032 | | | 885 |
United States | | | 1,773 | | | 217 |
| | | 14,708 | | | 8,214 |
| 16. | Property, plant and equipment |
(in KUSD) | | | Leasehold improve- ments | | | Laboratory equipment | | | Office equipment | | | Hardware | | | Total |
Cost | | | | | | | | | | | |||||
January 1, 2018 | | | 291 | | | 671 | | | 340 | | | 388 | | | 1,690 |
Additions | | | 226 | | | 327 | | | 277 | | | 114 | | | 944 |
Disposals and scrapping | | | — | | | — | | | (7) | | | — | | | (7) |
Exchange difference | | | (3) | | | (59) | | | (13) | | | (5) | | | (80) |
December 31, 2018 | | | 514 | | | 939 | | | 597 | | | 497 | | | 2,547 |
Additions | | | 17 | | | 131 | | | 93 | | | 118 | | | 359 |
Exchange difference | | | 2 | | | 41 | | | 8 | | | 3 | | | 54 |
December 31, 2019 | | | 533 | | | 1,111 | | | 698 | | | 618 | | | 2,960 |
| | | | | | | | | | | ||||||
Accumulated depreciation | | | | | | | | | | | |||||
January 1, 2018 | | | (70) | | | (168) | | | (136) | | | (174) | | | (548) |
Depreciation charge | | | (57) | | | (159) | | | (118) | | | (154) | | | (488) |
Disposals and scrapping | | | — | | | — | | | 7 | | | — | | | 7 |
Exchange difference | | | 1 | | | 17 | | | 2 | | | 2 | | | 22 |
December 31, 2018 | | | (126) | | | (310) | | | (245) | | | (326) | | | (1,007) |
Depreciation charge | | | (82) | | | (204) | | | (149) | | | (117) | | | (552) |
Exchange difference | | | (1) | | | (19) | | | (3) | | | (2) | | | (25) |
December 31, 2019 | | | (209) | | | (533) | | | (397) | | | (445) | | | (1,584) |
| | | | | | | | | | | ||||||
Net book amount | | | | | | | | | | | |||||
December 31, 2018 | | | 388 | | | 629 | | | 352 | | | 171 | | | 1,540 |
December 31, 2019 | | | 324 | | | 578 | | | 301 | | | 173 | | | 1,376 |
(in KUSD) | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 |
Research and development expenses | | | 407 | | | 330 |
General and administrative expenses | | | 145 | | | 158 |
| | | 552 | | | 488 |
| 17. | Leases |
(in KUSD) | | | December 31, 2019 | | | January 1, 2019 |
Properties (offices) | | | 4,820 | | | 5,399 |
Vehicles | | | 78 | | | 24 |
Total right-of-use assets | | | 4,898 | | | 5,423 |
(in KUSD) | | | December 31, 2019 | | | January 1, 2019 |
Lease liabilities (short-term) | | | 1,132 | | | 924 |
Lease liabilities (long-term) | | | 3,899 | | | 4,499 |
Total lease liabilities | | | 5,031 | | | 5,423 |
(in KUSD) | | | | | | | |||
Right-of-Use Assets | | | Offices | | | Vehicles | | | Total |
Cost | | | | | | | |||
January 1, 2019 | | | 5,399 | | | 24 | | | 5,423 |
Additions | | | 466 | | | 78 | | | 544 |
Disposals | | | — | | | (24) | | | (24) |
Exchange difference | | | 22 | | | — | | | 22 |
December 31, 2019 | | | 5,887 | | | 78 | | | 5,965 |
Accumulated depreciation | | | | | | | |||
January 1, 2019 | | | — | | | — | | | — |
Depreciation charge | | | (1,051) | | | (13) | | | (1,064) |
Disposals and scrapping | | | — | | | 13 | | | 13 |
Exchange difference | | | (16) | | | — | | | (16) |
December 31, 2019 | | | (1,067) | | | — | | | (1,067) |
| | | | | | | ||||
Net book amount | | | | | | | |||
January 1, 2019 | | | 5,399 | | | 24 | | | 5,423 |
December 31, 2019 | | | 4,820 | | | 78 | | | 4,898 |
(in KUSD) | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 |
Research and development expenses | | | 837 | | | — |
General and administrative expenses | | | 227 | | | — |
| | | 1,064 | | | — |
(in KUSD) | | | | | | | |||
Lease liabilities | | | Offices | | | Vehicles | | | Total |
January 1, 2019 | | | 5,399 | | | 24 | | | 5,423 |
Additions | | | 466 | | | 78 | | | 544 |
Disposals | | | — | | | (12) | | | (12) |
Cash outflow (including interest) | | | (1,130) | | | (13) | | | (1,143) |
Interest | | | 140 | | | 1 | | | 141 |
Exchange difference | | | 78 | | | — | | | 78 |
December 31, 2019 | | | 4,953 | | | 78 | | | 5,031 |
| | | | | | | ||||
Lease liabilities (short-term) | | | 1,114 | | | 18 | | | 1,132 |
Lease liabilities (long-term) | | | 3,839 | | | 60 | | | 3,899 |
Total lease liabilities | | | 4,953 | | | 78 | | | 5,031 |
| 18. | Intangible assets |
(in KUSD) | | | Licenses | | | Software | | | Total |
Cost | | | | | | | |||
January 1, 2018 | | | 6,009 | | | 44 | | | 6,053 |
Additions | | | 1,481 | | | 45 | | | 1,526 |
Exchange difference | | | — | | | (2) | | | (2) |
December 31, 2018 | | | 7,490 | | | 87 | | | 7,577 |
Additions | | | 1,731 | | | 59 | | | 1,790 |
Exchange difference | | | — | | | 1 | | | 1 |
December 31, 2019 | | | 9,221 | | | 147 | | | 9,368 |
Accumulated amortization | | | | | | | |||
January 1, 2018 | | | (626) | | | (26) | | | (652) |
Amortization charge | | | — | | | (25) | | | (25) |
Impairment loss | | | (227) | | | — | | | (227) |
Exchange difference | | | — | | | 1 | | | 1 |
December 31, 2018 | | | (853) | | | (50) | | | (903) |
Amortization charge | | | — | | | (30) | | | (30) |
Exchange difference | | | — | | | (1) | | | (1) |
December 31, 2019 | | | (853) | | | (81) | | | (934) |
| | | | | | | ||||
Net book amount | | | | | | | |||
December 31, 2018 | | | 6,637 | | | 37 | | | 6,674 |
December 31, 2019 | | | 8,368 | | | 66 | | | 8,434 |
(in KUSD) | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 |
Research and development expenses | | | 14 | | | 17 |
General and administrative expenses | | | 16 | | | 8 |
| | | 30 | | | 25 |
| - | An amount of KUSD 1,000 relating to a license agreement with a third party to acquire an antibody to be used in pre-clinical formalization, clinical testing, manufacturing and commercialization was capitalized as intangible assets. |
| - | An amount of KUSD 500 relating to a license agreement with a third party to use their technology to generate antibody-drug conjugates for up to five antibodies was capitalized as intangible assets. |
| - | An amount of KUSD 231 relating to a license agreement with a third party to use their technology for the production of antibodies was capitalized as intangible assets. |
| - | An amount of KUSD 231 relating to a license agreement with a third party to use their technology for the production of antibodies was capitalized as intangible assets. |
| - | An amount of KUSD 750 relating to a license agreement with a third party to use their technology to generate antibody-drug conjugates for up to five antibodies was capitalized as intangible assets. |
| - | An amount of KUSD 250 relating to a license agreement with a third party to use their technology to generate antibody-drug conjugates for up to five antibodies was capitalized as intangible assets. |
| - | An amount of KUSD 250 relating to a license agreement with a third party to acquire an antibody to be used in pre-clinical formalization, clinical testing, manufacturing and commercialization was capitalized as intangible assets. |
| - | Historical expenditure on clinical trials, future contractual commitments and internal budgets approved by the Board of Directors for ongoing and future trials; |
| - | Consideration of the progress of clinical trials, including obtaining primary endpoint readout data, discussions with regulatory authorities for new trials and enrollment status for ongoing clinical trials; |
| - | Consideration of market potential, supported where available by external market studies, and assessments of competitor products and product candidates. |
| 19a. | Financial instruments by class and by category |
(in KUSD) | | | Note | | | December 31, 2019 | | | December 31, 2018 |
Financial assets - financial assets at amortized cost | | | | | | | |||
Cash and cash equivalents | | | | | 115,551 | | | 138,807 | |
Trade accounts receivable | | | 13 | | | — | | | 192 |
Other current assets (excluding prepaid expenses) | | | 14 | | | 2,840 | | | 1,697 |
Other long-term assets | | | | | 368 | | | 264 | |
Total financial assets | | | | | 118,759 | | | 140,960 | |
| | | | | | |
(in KUSD) | | | Note | | | December 31, 2019 | | | December 31, 2018 |
Financial liabilities - financial liabilities at amortized cost | | | | | | | |||
Trade accounts payable | | | | | 3,329 | | | 6,750 | |
Accrued liabilities and other payables | | | 21 | | | 15,430 | | | 13,650 |
Lease liabilities | | | 17 | | | 5,031 | | | — |
Total financial liabilities | | | | | 23,790 | | | 20,400 | |
| | | | | | | ||||
Net financial position | | | | | 94,969 | | | 120,560 |
| 19b. | Credit quality of financial assets |
(in KUSD) | | | December 31, 2019 | | | December 31, 2018 |
Cash and cash equivalents | | | | | ||
A+ UBS | | | 51,983 | | | 71,493 |
A+ Credit Suisse | | | 62,652 | | | 66,066 |
A+ JP Morgan Chase | | | 916 | | | 1,248 |
| | | 115,551 | | | 138,807 |
| 20. | Deferred income taxes and tax credit |
(in KUSD) | | | December 31, 2019 | | | December 31, 2018 |
Deductible temporary differences, unused tax losses and unused tax credits for which no deferred tax assets have been recognized are attributable to the following: | | | | | ||
Taxes losses | | | 437,013 | | | 329,254 |
Unused tax credit | | | 10,765 | | | 6,009 |
Deductible temporary differences related to the retirement benefit plan | | | 2,684 | | | 1,386 |
Deductible temporary differences related to IFRS 16 | | | (39) | | | — |
Offset of recognized temporary differences related to intangible assets | | | (2,364) | | | (2,364) |
Total | | | 448,059 | | | 334,285 |
Years of expiry | | | December 31, 2019 | | | December 31, 2018 |
2019 | | | — | | | 6,209 |
2020 | | | 14,735 | | | 14,966 |
2021 | | | 19,889 | | | 20,200 |
2022 | | | 31,128 | | | 31,615 |
2023 | | | 38,441 | | | 39,042 |
Beyond 2024 | | | 332,820 | | | 217,222 |
| | | 437,013 | | | 329,254 |
Years of expiry | | | December 31, 2019 | | | December 31, 2018 |
2036 | | | 783 | | | 783 |
2037 | | | 1,839 | | | 1,839 |
2038 | | | 3,387 | | | 3,387 |
2039 | | | 4,756 | | | — |
| | | 10,765 | | | 6,009 |
| 21. | Accrued liabilities and other payables |
(in KUSD) | | | December 31, 2019 | | | December 31, 2018 |
Payroll and social charges | | | 5,726 | | | 4,939 |
Research and development costs | | | 8,922 | | | 8,124 |
Other | | | 782 | | | 587 |
| | | 15,430 | | | 13,650 |
| 22. | Pension obligations |
(in KUSD) | | | December 31, 2019 | | | December 31, 2018 |
Present value of defined benefit obligation for funded plan | | | 7,880 | | | 4,372 |
Fair value of plan assets | | | (5,196) | | | (2,986) |
Deficit of funded plan: liability on the balance sheet | | | 2,684 | | | 1,386 |
(in KUSD) | | | Present value of obligation | | | Fair value of plan assets | | | Total |
January 1, 2018 | | | 3,070 | | | (1,994) | | | 1,076 |
Defined benefit plan - pension costs: | | | | | | | |||
Current service cost | | | 527 | | | — | | | 527 |
Interest cost / (income) | | | 23 | | | (15) | | | 8 |
Defined benefit plan - pension costs | | | 550 | | | (15) | | | 535 |
Employee contributions | | | 208 | | | (208) | | | — |
Employer contributions | | | — | | | (416) | | | (416) |
Transfers from joiners' previous plans | | | 382 | | | (382) | | | — |
| | | 590 | | | (1,006) | | | (416) | |
Exchange differences | | | (12) | | | 10 | | | (2) |
Remeasurements: | | | | | | | |||
Change in financial assumptions | | | (99) | | | — | | | (99) |
Other actuarial losses | | | 295 | | | — | | | 295 |
Plan asset losses | | | — | | | 5 | | | 5 |
Exchange differences | | | (22) | | | 14 | | | (8) |
Remeasurements | | | 174 | | | 19 | | | 193 |
December 31, 2018 | | | 4,372 | | | (2,986) | | | 1,386 |
(in KUSD) | | | Present value of obligation | | | Fair value of plan assets | | | Total |
Defined benefit plan - pension costs: | | | | | | | |||
Current service cost | | | 769 | | | — | | | 769 |
Impact of plan changes | | | (319) | | | — | | | (319) |
Interest cost / (income) | | | 37 | | | (25) | | | 12 |
Defined benefit plan - pension costs | | | 487 | | | (25) | | | 462 |
Employee contributions | | | 257 | | | (257) | | | — |
Employer contributions | | | — | | | (515) | | | (515) |
Transfers from joiners' previous plans | | | 1,302 | | | (1,302) | | | — |
| | | 1,559 | | | (2,074) | | | (515) | |
Exchange differences | | | 74 | | | (69) | | | 5 |
Remeasurements: | | | | | | | |||
Change in financial assumptions | | | 686 | | | — | | | 686 |
Other actuarial losses | | | 609 | | | — | | | 609 |
Plan asset gains | | | — | | | (2) | | | (2) |
Exchange differences | | | 93 | | | (40) | | | 53 |
Remeasurements | | | 1,388 | | | (42) | | | 1,346 |
December 31, 2019 | | | 7,880 | | | (5,196) | | | 2,684 |
| | | 2019 | | | 2018 | |
Discount rate | | | 0.20% | | | 0.85% |
Interest credited on savings accounts | | | 0.20% | | | 1.00% |
Future salary increases | | | 1.50% | | | 1.50% |
Future pension increases | | | 0.00% | | | 0.00% |
| | | 2019 | | | 2018 | |
Male | | | 22.61 | | | 22.50 |
Female | | | 25.64 | | | 25.53 |
2019 | | | Increase in assumption | | | Impact on defined benefit obligation and service cost | | | Decrease in assumption | | | Impact on defined benefit obligation and service cost |
Discount rate | | | 0.25% | | | (5.40%) | | | (0.25%) | | | 5.90% |
Future salary increases | | | 0.50% | | | 0.90% | | | (0.50%) | | | (0.90%) |
Interest credited on savings accounts | | | 0.50% | | | 3.20% | | | (0.50%) | | | (3.00%) |
Future pension increases | | | 0.50% | | | 7.00% | | | (0.50%) | | | (6.30%) |
2018 | | | Increase in assumption | | | Impact on defined benefit obligation and service cost | | | Decrease in assumption | | | Impact on defined benefit obligation and service cost |
Discount rate | | | 0.25% | | | (5.40%) | | | (0.25%) | | | 5.90% |
Future salary increases | | | 0.50% | | | 1.00% | | | (0.50%) | | | (1.00%) |
Interest credited on savings accounts | | | 0.50% | | | 3.20% | | | (0.50%) | | | (3.10%) |
Future pension increases | | | 0.50% | | | 6.20% | | | (0.50%) | | | (5.60%) |
| | | December 31, 2019 | | | December 31, 2018 | |
Cash | | | 155 | | | 65 |
Bonds | | | 3,065 | | | 1,919 |
Shares | | | 329 | | | 147 |
Real estates and mortgages | | | 1,303 | | | 720 |
Alternative investments | | | 344 | | | 135 |
| | | 5,196 | | | 2,986 |
(in KUSD) | | | 2019 | | | 2018 |
January 1 | | | (1,043) | | | (850) |
Remeasurements of defined benefit pension plan | | | (1,346) | | | (193) |
December 31 | | | (2,389) | | | (1,043) |
| 23. | Share-based compensation expense |
| | | 2019 | | | 2018 | |||||||
| | | Average strike price in USD per share | | | Number of awards | | | Average strike price in USD per share | | | Number of awards | |
At the beginning of the year | | | 12.5 | | | 1,982,375 | | | 11.8 | | | 1,882,125 |
Granted | | | 26.6 | | | 226,666 | | | 25.9 | | | 100,250 |
Forfeited | | | 9.8 | | | (183,509) | | | — | | | — |
At the end of the year | | | 14.3 | | | 2,025,532 | | | 12.5 | | | 1,982,375 |
Grant date | | | Expiry date | | | Strike price in USD | | | Awards December 31, 2019 | | | Awards December 31, 2018 |
Feb - Sep 2014 | | | 2024 | | | 9.3 | | | 602,375 | | | 602,375 |
Mar - Jul 2015 | | | 2025 | | | 9.3 | | | 144,875 | | | 144,875 |
Oct - Nov 2015 | | | 2025 | | | 11.2 | | | 11,875 | | | 11,875 |
Jan - Jul 2016 | | | 2026 | | | 11.2 | | | 151,375 | | | 151,375 |
Jan - Oct 2017 | | | 2027 | | | 13.4 | | | 938,250 | | | 938,250 |
Nov - Dec 2017 | | | 2027 | | | 25.9 | | | 33,375 | | | 33,375 |
Jan - Dec 2018 | | | 2028 | | | 25.9 | | | 100,250 | | | 100,250 |
Jan - Apr 2019 | | | 2029 | | | 25.9 | | | 155,062 | | | |
Jul - Sep 2019 | | | 2029 | | | 28.0 | | | 71,604 | | | |
Jul 2019 forfeited | | | | | | | (183,509) | | | |||
Total | | | | | | | 2,025,532 | | | 1,982,375 | ||
Weighted average remaining contractual life of awards outstanding at end of period | | | | | | | 6.64 years | | | 7.17 years |
| | | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 | ||
a) weighted average share price | | | in USD | | | 0.80 to 27.50 | | | 0.79 to 1.91 |
b) strike price | | | in USD | | | 26.00 to 28.00 | | | 25.94 |
c) expected volatility | | | in % | | | 201.9 to 4,612.8 | | | 146.4 to 201.9 |
d) award life | | | in # of years | | | 0.08 to 1.02 | | | 1.02 to 1.75 |
e) expected dividends | | | in % | | | 0 | | | 0 |
f) risk-free interest rate | | | in % | | | 2.08 to 2.57 | | | 1.84 to 2.57 |
| | | 2019 | | | 2018 | |||||||
| | | Average strike price in USD per share | | | Number of awards | | | Average strike price in USD per share | | | Number of awards | |
At the beginning of the year | | | 2.7 | | | 2,762,500 | | | 2.7 | | | 2,312,500 |
Granted | | | 2.5 | | | 584,460 | | | 2.6 | | | 450,000 |
Forfeited | | | — | | | — | | | — | | | — |
At the end of the year | | | 2.6 | | | 3,346,960 | | | 2.7 | | | 2,762,500 |
Grant date | | | Expiry date | | | Strike price in USD | | | Awards December 31, 2019 | | | Awards December 31, 2018 |
May 2014 | | | 2024 | | | 2.8 | | | 63,750 | | | 63,750 |
Sep 2014 | | | 2024 | | | 2.8 | | | 102,500 | | | 102,500 |
May 2015 | | | 2025 | | | 2.7 | | | 1,047,375 | | | 1,047,375 |
Sep 2015 | | | 2025 | | | 2.7 | | | 160,500 | | | 160,500 |
May 2016 | | | 2026 | | | 2.7 | | | 101,125 | | | 101,125 |
May 2016 | | | 2026 | | | 2.7 | | | 160,500 | | | 160,500 |
Oct 2016 | | | 2026 | | | 2.6 | | | 112,500 | | | 112,500 |
Dec 2016 | | | 2026 | | | 2.6 | | | 564,250 | | | 564,250 |
Jun 2018 | | | 2028 | | | 2.6 | | | 37,500 | | | 37,500 |
Dec 2018 | | | 2028 | | | 2.6 | | | 412,500 | | | 412,500 |
Jan 2019 | | | 2029 | | | 2.6 | | | 75,000 | | | |
Dec 2019 | | | 2029 | | | 2.5 | | | 509,460 | | | |
Total | | | | | | | 3,346,960 | | | 2,762,500 | ||
Weighted average remaining contractual life of awards outstanding at end of period | | | | | | | 6.97 years | | | 7.37 years |
| | | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 | ||
a) weighted average share price | | | in USD | | | 0.51 to 16.31 | | | 0.50 to 1.25 |
b) strike price | | | in USD | | | 2.50 to 2.56 | | | 2.58 to 2.63 |
c) expected volatility | | | in % | | | 176.6 to 233.1 | | | 170.0 to 233.1 |
d) award life | | | in # of years | | | 0.83 to 1.29 | | | 0.83 to 1.33 |
e) expected dividends | | | in % | | | 0 | | | 0 |
f) risk-free interest rate | | | in % | | | 1.55 to 2.60 | | | 2.32 to 2.60 |
| | | 2019 | | | 2018 | |||||||
| | | Average strike price in USD per share | | | Number of awards | | | Average strike price in USD per share | | | Number of awards | |
At the beginning of the year | | | — | | | — | | | — | | | — |
Granted | | | 18.8 | | | 1,020,434 | | | — | | | — |
Forfeited | | | — | | | — | | | — | | | — |
At the end of the year | | | 18.8 | | | 1,020,434 | | | — | | | — |
Grant date | | | Expiry date | | | Strike price in USD | | | Awards December 31, 2019 | | | Awards December 31, 2018 |
Dec 2019 | | | 2029 | | | 18.8 | | | 1,020,434 | | | |
Total | | | | | | | 1,020,434 | | | — | ||
Weighted average remaining contractual life of awards outstanding at end of period | | | | | | | 9.96 years | | | — |
| | | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 | ||
a) weighted average share price | | | in USD | | | 16.31 | | | — |
b) strike price | | | in USD | | | 18.75 | | | — |
c) expected volatility | | | in % | | | 176.6 | | | — |
d) award life | | | in # of years | | | 5.65 | | | — |
e) expected dividends | | | in % | | | 0 | | | — |
f) risk-free interest rate | | | in % | | | 1.67 | | | — |
(in KUSD) | | | 2019 | | | 2018 |
January 1 | | | 6,745 | | | 6,276 |
Share Purchase Plan 2013 | | | — | | | — |
Incentive Plan 2014 | | | 437 | | | 108 |
Share Purchase Plan 2016 | | | 332 | | | 361 |
Equity Incentive Plan 2019 | | | 348 | | | — |
December 31 | | | 7,862 | | | 6,745 |
| 24. | Share capital |
| | | Number of shares issued Common | | | Number of shares issued Class B | | | Number of shares issued Class C | | | Number of shares issued Class D | | | Number of shares issued Class E | | | Number of shares issued Total | |
January 1, 2018 | | | 9,487,500 | | | 14,262,500 | | | 8,075,000 | | | 7,837,500 | | | 7,712,500 | | | 47,375,000 |
Issuance of share capital / capital contributions | | | 450,000 | | | — | | | — | | | — | | | — | | | 450,000 |
December 31, 2018 | | | 9,937,500 | | | 14,262,500 | | | 8,075,000 | | | 7,837,500 | | | 7,712,500 | | | 47,825,000 |
Issuance of share capital / capital contributions | | | 1,825,000 | | | — | | | — | | | — | | | 3,687,500 | | | 5,512,500 |
December 31, 2019 | | | 11,762,500 | | | 14,262,500 | | | 8,075,000 | | | 7,837,500 | | | 11,400,000 | | | 53,337,500 |
of which, treasury shares: | | | 1,240,540 | | | | | | | | | | |
| | | Share capital (in KUSD) | | | Share premium (in KUSD) | | | Treasury shares (in KUSD) | | | Total (in KUSD) | |
January 1, 2018 | | | 397 | | | 452,296 | | | — | | | 452,693 |
Issuance of share capital /capital contributions | | | 4 | | | (28) | | | — | | | (24) |
December 31, 2018 | | | 401 | | | 452,268 | | | — | | | 452,669 |
Issuance of share capital /capital contributions | | | 171 | | | 101,443 | | | — | | | 101,614 |
Change in par value | | | 3,789 | | | (3,789) | | | | | — | |
Treasury shares - additions | | | — | | | — | | | (141) | | | (141) |
Treasury shares - disposals | | | — | | | — | | | 41 | | | 41 |
December 31, 2019 | | | 4,361 | | | 549,922 | | | (100) | | | 554,183 |
| 1. | First, to the Class E shareholders, an amount equal to the total price paid for the Class E shares, plus an 8% per annum return; |
| 2. | Second, to the Class D shareholders, an amount equal to the total price paid for the Class D shares, plus an 8% per annum return; |
| 3. | Third, to the Class C shareholders, an amount equal to the total price paid for the Class C shares; |
| 4. | Fourth, pari passu, to the Class B shareholders, an amount equal to the total price paid for the Class B shares, plus an 8% per annum return, and, to the Class C shareholders, an 8% per annum return on the total price paid for the Class C shares; and |
| 5. | Thereafter, to all shareholders, the remaining proceeds in proportion to the nominal value of their shares. |
| | | 2019 | | | 2018 | |||||||||||||
| | | Price (in USD) | | | Number of treasury shares | | | Value (in KUSD) | | | Price (in USD) | | | Number of treasury shares | | | Value (in KUSD) | |
At the beginning of the year | | | — | | | — | | | — | | | — | | | — | | | — |
Additions | | | 0.08 | | | 1,750,000 | | | 141 | | | — | | | — | | | — |
Disposals | | | 0.08 | | | (509,460) | | | (41) | | | — | | | — | | | — |
At the end of the year | | | 0.08 | | | 1,240,540 | | | 100 | | | — | | | — | | | — |
| 25. | Commitments |
(in KUSD): | | | | | ||
R&D Phase | | | December 31, 2019 | | | December 31, 2018 |
Pre-clinical | | | 192,290 | | | 275,602 |
Phase I | | | 116,115 | | | 49,959 |
Phase II | | | 15,568 | | | 5,569 |
| | | 323,973 | | | 331,130 |
(in KUSD) | | | December 31, 2018 |
Not later than 1 year | | | 1,084 |
Later than 1 year and not later than 5 years | | | 3,163 |
More than 5 years | | | 131 |
| | | 4,378 |
| 26. | Contingent liabilities |
| 27. | Related parties |
(in KUSD) | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 |
Salaries and other short-term employee benefits | | | 5,364 | | | 4,512 |
Pension costs | | | 407 | | | 299 |
Share-based compensation expenses | | | 396 | | | 341 |
Other compensation | | | 73 | | | 183 |
| | | 6,240 | | | 5,335 |
| 28. | Loss per share |
Year Ended December 31, 2019 | | | Common Shares | | | Class B Preferred Shares | | | Class C Preferred Shares | | | Class D Preferred Shares | | | Class E Preferred Shares |
Net loss attributable to shareholders (in KUSD) | | | (22,137) | | | (33,713) | | | (19,087) | | | (18,526) | | | (23,021) |
Weighted average number of shares in issue | | | 9,365,406 | | | 14,262,500 | | | 8,075,000 | | | 7,837,500 | | | 9,739,555 |
Basic and diluted loss per share (in USD) | | | (2.36) | | | (2.36) | | | (2.36) | | | (2.36) | | | (2.36) |
Year Ended December 31, 2018 | | | Common Shares | | | Class B Preferred Shares | | | Class C Preferred Shares | | | Class D Preferred Shares | | | Class E Preferred Shares |
Net loss attributable to shareholders (in KUSD) | | | (23,014) | | | (37,676) | | | (21,330) | | | (20,703) | | | (20,373) |
Year Ended December 31, 2018 | | | Common Shares | | | Class B Preferred Shares | | | Class C Preferred Shares | | | Class D Preferred Shares | | | Class E Preferred Shares |
Weighted average number of shares in issue | | | 8,712,500 | | | 14,262,500 | | | 8,075,000 | | | 7,837,500 | | | 7,712,500 |
Basic and diluted loss per share (in USD) | | | (2.64) | | | (2.64) | | | (2.64) | | | (2.64) | | | (2.64) |
| 29. | Foreign currency exchange rate |
USD / GBP | | | | | Year ended December 31, 2019 | | | Year ended December 31, 2018 | |
Closing rate, GBP 1 | | | USD | | | 1.31858 | | | 1.26902 |
Weighted average exchange rate, GBP 1 | | | USD | | | 1.27468 | | | 1.35009 |
| Events after the reporting date |
| Item 6. | Indemnification of Directors and Officers |
| Item 7. | Recent Sales of Unregistered Securities |
Name or Class of Purchasers | | | Date of Issuance | | | Title of Securities | | | Number of Securities | | | Consideration (in USD thousands) |
Directors, officers, employees and consultants | | | December 16, 2019 | | | Common shares | | | 636,825 | | | 1,274 |
| | | | | | | | | |||||
A.T. Holdings II Sàrl (for the creation of treasury shares to settle share grants and equity-linked instruments for directors, officers employees and consultants) | | | September 19, 2019 | | | Class A common shares | | | 140 | | | 141 |
| | | | | | | | | |||||
Various private equity investment funds, institutional investors and other persons | | | June 7, 2019 June 14, 2019 July 5, 2019 | | | Class E preferred shares | | | 295 | | | 103,250 |
| | | | | | | | |
Name or Class of Purchasers | | | Date of Issuance | | | Title of Securities | | | Number of Securities | | | Consideration (in USD thousands) |
Directors and officers | | | June 29, 2018 December 14, 2018 February 6, 2019 | | | Class A common shares | | | 42 | | | 1,314 |
| | ||||||||||||
Various private equity investment funds, institutional investors and other persons | | | October 12, 2017 October 30, 2017 November 16, 2017 | | | Class E preferred shares | | | 617 | | | 200,088 |
| Item 8. | Exhibits and Financial Statement Schedules |
| Item 9. | Undertakings |
| (a) | for purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective; |
| (b) | for the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof; |
| (c) | insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer, or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless, in the opinion of its counsel, the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question of whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue; and |
| (d) | to provide to the underwriters at the closing specified in the underwriting agreement, certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser. |
| | | Form of Underwriting Agreement | |
| | | Form of Amended and Restated Articles of Association of ADC Therapeutics SA, to be in effect immediately prior to the consummation of this offering | |
| | | Opinion of Homburger AG, Swiss counsel of ADC Therapeutics SA, as to the validity of the common shares | |
| | | Second Amended and Restated License Agreement among ADC Products (UK) Limited, ADC Therapeutics SA and MedImmune Limited, dated May 9, 2016 | |
| | | Amendment #1 to the Second Amended and Restated License Agreement among ADC Products (UK) Limited, ADC Therapeutics SA and MedImmune Limited, dated September 19, 2018 | |
| | | Collaboration and License Agreement between ADC Therapeutics Sarl and Genmab A/S, dated June 14, 2013 | |
| | | Amendment to the Collaboration and License Agreement between ADC Therapeutics Sarl and Genmab A/S, dated November 20, 2013 | |
| | | Second Amendment to the Collaboration and License Agreement between ADC Therapeutics SA and Genmab A/S, dated April 16, 2020 | |
| | | Lease Relating to Suite 5, 1st Floor, The Queen Mary BioEnterprises Innovation Centre between Queen Mary Bioenterprises Limited and ADC Therapeutics (UK) Limited, dated September 14, 2017 | |
| | | Lease Relating to Lab 11b, Suite 11 Write Up Space and Suite 12, 1st Floor, The Queen Mary BioEnterprises Innovation Centre between Queen Mary Bioenterprises Limited and ADC Therapeutics (UK) Limited, dated December 20, 2017 | |
| | | Counterpart Lease Relating to Suite 8, First Floor, The Queen Mary BioEnterprises Innovation Centre between Queen Mary Bioenterprises Limited and ADC Therapeutics (UK) Limited, dated July 9, 2018 | |
| | | Deed of Variation Relating to Lease of Suite 5, 1st Floor, The Queen Mary BioEnterprises Innovation Centre between Queen Mary Bioenterprises Limited and ADC Therapeutics (UK) Limited, dated July 1, 2019 | |
| | | Deed of Variation Relating to Lease of Lab 11b, Suite 11 Write Up Space and Suite 12, 1st Floor, The Queen Mary BioEnterprises Innovation Centre between Queen Mary Bioenterprises Limited and ADC Therapeutics (UK) Limited, dated July 1, 2019 | |
| | | Deed of Variation Relating to Lease of Suite 8, First Floor, The Queen Mary BioEnterprises Innovation Centre between Queen Mary Bioenterprises Limited and ADC Therapeutics (UK) Limited, dated July 1, 2019 | |
| | | Form of Indemnity Agreement with directors and officers entered into in connection with this offering | |
| | | Form of Purchase and Shareholders Agreement relating to the 2013 Share Purchase Plan (including form of promissory note included in Schedule A thereto) | |
| | | ADC Therapeutics Incentive Plan, between ADC Therapeutics SA and the parties named therein, dated May 1, 2014 as amended and restated as of October 1, 2015 | |
| | | 2016 Share Purchase Plan, between ADC Therapeutics Ltd and the parties named therein, dated November 18, 2016 (including form of promissory note including in Annex 3A thereto) | |
| | | Form of 2019 Equity Incentive Plan | |
| | | Facility Agreement among ADC Therapeutics SA, the other Loan Parties party thereto, the Lenders and Deerfield Partners, L.P., as agent for itself and the Secured Parties thereto, dated April 24, 2020 | |
| | | Form of Senior Secured Convertible Note | |
| | | Form of Registration Rights Agreement between ADC Therapeutics SA and Deerfield Partners, L.P. and Deerfield Private Design Fund IV, L.P., to be entered into in connection with this offering | |
| | | List of subsidiaries | |
| | | Consent of PricewaterhouseCoopers SA | |
| | | Consent of Homburger AG, Swiss counsel of ADC Therapeutics SA (included in Exhibit 5.1) | |
| | | Powers of attorney (included on signature page to the registration statement) | |
| | | Consent of Jennifer Creel to be named as Chief Financial Officer nominee |
| * | To be filed by amendment. |
| ** | Previously filed. |
| # | Portions of this exhibit have been omitted because they are both (i) not material and (ii) would likely cause competitive harm to the Company if publicly disclosed. |
| | | ADC THERAPEUTICS SA | |||||||
| | | | | | | ||||
| | | By: | | | /s/ Christopher Martin | ||||
| | | | | Name: | | | Christopher Martin | ||
| | | | | Title: | | | Chief Executive Officer | ||
Name | | | | | Title | |
| | | | | |||
/s/ Christopher Martin | | | | | Chief Executive Officer and Director (principal executive officer) | |
Christopher Martin | | | | |||
| | | | | |||
/s/ Michael Forer | | | | | Chief Financial Officer and Vice Chairman of the Board of Directors (principal financial officer and principal accounting officer) | |
Michael Forer | | | | |||
| | | | ||||
* | | | | | Chairman of the Board of Directors | |
Ron Squarer | | | | |||
| | | | | |||
* | | | | | Director | |
Peter B. Corr | | | | |||
| | | | | |||
* | | | | | Director | |
Stephen Evans-Freke | | | | |||
| | | | | |||
* | | | | | Director | |
Peter Hug | | | | |||
| | | | | |||
* | | | | | Director | |
Thomas Pfisterer | | | | |||
| | | | | |||
* | | | | | Director | |
Thomas M. Rinderknecht | | | | |||
| | | | | |||
* | | | | | Director | |
Tyrell J. Rivers | | | | |||
| | | | | |||
* | | | | | Director | |
Victor Sandor | | | | |||
| | | | | |||
* | | | | | Director | |
Jacques Theurillat | | | | |||
| | | | | |||
/s/ Jay Feingold | | | | | Authorized Representative in the United States | |
Jay Feingold | | |||||
ADC Therapeutics America, Inc. | | | |
* By: | | | /s/ Christopher Martin | | | |
| | | Christopher Martin | | | ||
| | | Attorney-in-Fact | | |