UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-Q
(Mark One)
| | | |
| þ | | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934. |
For the quarterly period ended March 31, 2006
OR
| | | |
| o | | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934. |
For the transition period from to .
Commission File No. 1-15803
AVANIR PHARMACEUTICALS
(Exact name of registrant as specified in its charter)
| | | |
California
(State or other jurisdiction of
incorporation or organization) | | 33-0314804
(I.R.S. Employer Identification No.) |
| | | |
11388 Sorrento Valley Road, San Diego, California
(Address of principal executive offices) | | 92121
(Zip Code) |
(858) 622-5200
(Registrant’s telephone number, including area code)
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities and Exchange Act of 1934 during the preceding 12 months (or for such shorter periods that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YESþ NOo
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
| | | | |
| Large Accelerated Filer o | | Accelerated Filer þ | | Non-Accelerated Filer o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
YESo NOþ
As of May 8, 2006, the registrant had 31,675,721 shares of common stock issued and outstanding.
PART I. FINANCIAL INFORMATION
Item 1. FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Shareholders
Avanir Pharmaceuticals
We have reviewed the accompanying condensed consolidated balance sheet ofAvanir Pharmaceuticals and subsidiaries (the “Company”) as of March 31, 2006 and the related condensed consolidated statements of operations for the three-month and six-month periods ended March 31, 2006 and 2005 and of cash flows for the six-month periods ended March 31, 2006 and 2005. These interim financial statements are the responsibility of the Company’s management.
We conducted our reviews in accordance with the standards of the Public Company Accounting Oversight Board (United States). A review of interim financial information consists principally of applying analytical procedures and making inquiries of persons responsible for financial and accounting matters. It is substantially less in scope than an audit conducted in accordance with the standards of the Public Company Accounting Oversight Board (United States), the objective of which is the expression of an opinion regarding the financial statements taken as a whole. Accordingly, we do not express such an opinion.
Based on our reviews, we are not aware of any material modifications that should be made to such condensed consolidated interim financial statements for them to be in conformity with accounting principles generally accepted in the United States of America.
We have previously audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheet ofAvanir Pharmaceuticals and subsidiaries as of September 30, 2005, and the related consolidated statements of operations, shareholders’ equity and comprehensive loss, and cash flows for the year then ended (not presented herein); and in our report dated December 14, 2005, we expressed an unqualified opinion on those consolidated financial statements. In our opinion, the information set forth in the accompanying condensed consolidated balance sheet as of September 30, 2005 is fairly stated, in all material respects, in relation to the consolidated balance sheet from which it has been derived.
DELOITTE & TOUCHE LLP
San Diego, California
May 10, 2006
3
Avanir Pharmaceuticals
CONDENSED CONSOLIDATED BALANCE SHEETS (UNAUDITED)
| | | | | | | | | |
| | | March 31, | | | September 30, | |
| | | 2006 | | | 2005 | |
ASSETS | | | | | | | | |
| Current assets: | | | | | | | | |
| Cash and cash equivalents | | $ | 10,912,580 | | | $ | 8,620,143 | |
| Short-term investments in securities | | | 38,738,504 | | | | 14,215,005 | |
| Receivables, net | | | 1,482,674 | | | | 1,169,654 | |
| Inventory | | | 271,375 | | | | 27,115 | |
| Prepaid expenses | | | 1,566,671 | | | | 2,370,801 | |
| | | | | | | |
| Total current assets | | | 52,971,804 | | | | 26,402,718 | |
| Investments in securities | | | 2,446,575 | | | | 3,845,566 | |
| Restricted investments in securities | | | 856,597 | | | | 856,872 | |
| Property and equipment, net | | | 6,111,449 | | | | 6,004,527 | |
| Intangible assets, net | | | 3,726,164 | | | | 3,665,086 | |
| Long-term inventory | | | 522,508 | | | | 347,424 | |
| Other assets | | | 350,125 | | | | 279,797 | |
| | | | | | | |
| TOTAL ASSETS | | $ | 66,985,222 | | | $ | 41,401,990 | |
| | | | | | | |
LIABILITIES AND SHAREHOLDERS’ EQUITY | | | | | | | | |
| Current liabilities: | | | | | | | | |
| Accounts payable | | $ | 3,252,387 | | | $ | 6,751,781 | |
| Accrued expenses and other liabilities | | | 7,220,922 | | | | 4,094,295 | |
| Accrued compensation and payroll taxes | | | 1,223,772 | | | | 1,272,231 | |
| Current portion of deferred revenue | | | 2,316,118 | | | | 1,970,989 | |
| Current portion of notes payable | | | 333,317 | | | | 317,667 | |
| Current portion of capital lease obligations | | | 22,816 | | | | 26,305 | |
| | | | | | | |
| Total current liabilities | | | 14,369,332 | | | | 14,433,268 | |
| Deferred revenue, net of current portion | | | 16,683,843 | | | | 17,187,221 | |
| Notes payable, net of current portion | | | 466,625 | | | | 637,285 | |
| Capital lease obligations, net of current portion | | | — | | | | 9,337 | |
| | | | | | | |
| Total liabilities | | | 31,519,800 | | | | 32,267,111 | |
| | | | | | | |
COMMITMENTS AND CONTINGENCIES(Note 14) | | | | | | | | |
| | | | | | | | | |
| SHAREHOLDERS’ EQUITY: | | | | | | | | |
| Preferred stock — no par value, 10,000,000 shares authorized, no shares issued and outstanding as of March 31, 2006 and September 30, 2005 | | | — | | | | — | |
| Common stock — no par value, Class A, 200,000,000 shares authorized, 31,653,236 and 27,341,732 shares issued and outstanding as of March 31, 2006 and September 30, 2005, respectively (Note 2) | | | 209,862,878 | | | | 167,738,303 | |
| Unearned compensation | | | — | | | | (3,477,144 | ) |
| Accumulated deficit | | | (174,092,311 | ) | | | (155,012,466 | ) |
| Accumulated other comprehensive loss | | | (305,145 | ) | | | (113,814 | ) |
| | | | | | | |
| Total shareholders’ equity | | | 35,465,422 | | | | 9,134,879 | |
| | | | | | | |
| TOTAL LIABILITIES AND SHAREHOLDERS’ EQUITY | | $ | 66,985,222 | | | $ | 41,401,990 | |
| | | | | | | |
See notes to condensed consolidated financial statements.
4
AVANIR Pharmaceuticals
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS (UNAUDITED)
| | | | | | | | | | | | | | | | | |
| | | Three Months Ended | | | Six Months Ended | |
| | | March 31, | | | March 31, | | |
| | | 2006 | | | 2005 | | | 2006 | | | 2005 | |
REVENUES: | | | | | | | | | | | | | | | | |
| Research and development services | | $ | 1,956,565 | | | $ | — | | | $ | 4,434,583 | | | $ | — | |
| Royalties and sales of royalty rights | | | 427,259 | | | | 419,816 | | | | 1,009,304 | | | | 930,130 | |
| Government research grants | | | 75,862 | | | | 124,917 | | | | 160,687 | | | | 285,568 | |
| Licenses | | | 9,342 | | | | 100,000 | | | | 5,009,342 | | | | 300,000 | |
| Product sales | | | — | | | | — | | | | — | | | | 17,400 | |
| | | | | | | | | | | | | |
| Total revenues | | | 2,469,028 | | | | 644,733 | | | | 10,613,916 | | | | 1,533,098 | |
| | | | | | | | | | | | | |
OPERATING EXPENSES: | | | | | | | | | | | | | | | | |
| Research and development | | | 7,919,847 | | | | 11,428,170 | | | | 17,283,249 | | | | 16,482,411 | |
| Selling, general and administrative | | | 8,492,115 | | | | 3,372,183 | | | | 13,260,858 | | | | 6,323,186 | |
| Cost of product sales | | | — | | | | — | | | | — | | | | 3,102 | |
| | | | | | | | | | | | | |
| Total operating expenses | | | 16,411,962 | | | | 14,800,353 | | | | 30,544,107 | | | | 22,808,699 | |
| | | | | | | | | | | | | |
LOSS FROM OPERATIONS | | | (13,942,934 | ) | | | (14,155,620 | ) | | | (19,930,191 | ) | | | (21,275,601 | ) |
| Interest income | | | 564,422 | | | | 125,147 | | | | 892,588 | | | | 246,979 | |
| Interest expense | | | (21,403 | ) | | | (25,324 | ) | | | (44,841 | ) | | | (46,945 | ) |
| Other income (expense), net | | | (5,492 | ) | | | 4,850 | | | | 5,020 | | | | (62,071 | ) |
| | | | | | | | | | | | | |
LOSS BEFORE INCOME TAXES | | | (13,405,407 | ) | | | (14,050,947 | ) | | | (19,077,424 | ) | | | (21,137,638 | ) |
| Provision for income taxes | | | (4 | ) | | | — | | | | (2,421 | ) | | | (1,898 | ) |
| | | | | | | | | | | | | |
NET LOSS | | $ | (13,405,411 | ) | | $ | (14,050,947 | ) | | $ | (19,079,845 | ) | | $ | (21,139,536 | ) |
| | | | | | | | | | | | | |
NET LOSS PER SHARE: | | | | | | | | | | | | | | | | |
| BASIC AND DILUTED (Note 2) | | $ | (0.43 | ) | | $ | (0.57 | ) | | $ | (0.64 | ) | | $ | (0.87 | ) |
| | | | | | | | | | | | | |
WEIGHTED AVERAGE NUMBER OF COMMON SHARES OUTSTANDING: | | | | | | | | | | | | | | | | |
| BASIC AND DILUTED (Note 2) | | | 31,086,874 | | | | 24,565,682 | | | | 29,819,338 | | | | 24,261,024 | |
| | | | | | | | | | | | | |
See notes to condensed consolidated financial statements.
5
AVANIR Pharmaceuticals
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS (UNAUDITED)
| | | | | | | | | |
| | | Six Months Ended March 31, | |
| | | 2006 | | | 2005 | |
OPERATING ACTIVITIES: | | | | | | | | |
| Net loss | | $ | (19,079,845 | ) | | $ | (21,139,536 | ) |
| Adjustments to reconcile net loss to net cash used for operating activities: | | | | | | | | |
| Depreciation and amortization | | | 965,679 | | | | 913,363 | |
| Share-based compensation expense | | | 1,219,733 | | | | 5,402 | |
| Issuance of common stock in connection with the acquisition of additional contractual rights to Neurodex™ | | | — | | | | 5,300,000 | |
| Other-than-temporary impairment of investment | | | — | | | | 78,012 | |
| Loss on disposal of assets | | | 8,994 | | | | 2,827 | |
| Intangible assets abandoned and impaired | | | 134,405 | | | | 177,965 | |
| Changes in operating assets and liabilities: | | | | | | | | |
| Receivables | | | (313,020 | ) | | | (81,118 | ) |
| Inventory | | | (419,344 | ) | | | 3,102 | |
| Prepaid expenses and other assets | | | 723,863 | | | | (611,219 | ) |
| Accounts payable | | | (3,476,650 | ) | | | (253,261 | ) |
| Accrued expenses and other liabilities | | | 3,116,777 | | | | 1,281,301 | |
| Accrued compensation and payroll taxes | | | (48,459 | ) | | | 90,531 | |
| Deferred revenue | | | (158,249 | ) | | | (1,028,145 | ) |
| | | | | | | |
| Net cash used for operating activities | | | (17,326,116 | ) | | | (15,260,776 | ) |
| | | | | | | |
INVESTING ACTIVITIES: | | | | | | | | |
| Investments in securities | | | (46,165,564 | ) | | | (4,339,986 | ) |
| Proceeds from sales and maturities of investments in securities | | | 22,850,000 | | | | 7,914,365 | |
| Patent costs | | | (328,672 | ) | | | (551,777 | ) |
| Purchases of property and equipment | | | (951,362 | ) | | | (151,289 | ) |
| | | | | | | |
| Net cash (used for) provided by investing activities. | | | (24,595,598 | ) | | | 2,871,313 | |
| | | | | | | |
FINANCING ACTIVITIES: | | | | | | | | |
| Proceeds from issuance of common stock, net | | | 44,381,987 | | | | 7,196,498 | |
| Proceeds from issuance of notes payable | | | — | | | | 136,438 | |
| Payments on notes and capital lease obligations | | | (167,836 | ) | | | (231,070 | ) |
| | | | | | | |
| Net cash provided by financing activities | | | 44,214,151 | | | | 7,101,866 | |
| | | | | | | |
| Net increase (decrease) in cash and cash equivalents | | | 2,292,437 | | | | (5,287,597 | ) |
| Cash and cash equivalents at beginning of period | | | 8,620,143 | | | | 13,494,083 | |
| | | | | | | |
| Cash and cash equivalents at end of period | | $ | 10,912,580 | | | $ | 8,206,486 | |
| | | | | | | |
SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION: | | | | | | | | |
| Interest paid | | $ | 44,841 | | | $ | 46,945 | |
| Income taxes paid | | $ | 2,421 | | | $ | 1,898 | |
NONCASH INVESTING AND FINANCING ACTIVITIES: | | | | | | | | |
| Accounts payable and accrued expenses for purchases of property and equipment | | $ | 229,319 | | | $ | 93,532 | |
| Elimination of unearned compensation against common stock | | $ | 3,477,144 | | | $ | — | |
See notes to condensed consolidated financial statements.
6
Avanir Pharmaceuticals
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
1. BASIS OF PRESENTATION
Avanir Pharmaceuticals (“Avanir,” “we,” or the “Company”) prepared the accompanying unaudited condensed consolidated financial statements in accordance with the rules and regulations of the Securities and Exchange Commission (“SEC”) including the instructions to Form 10-Q and Rule 10-01 of Regulation S-X. Such rules and regulations allow us to condense and omit certain information and footnote disclosures normally included in audited financial statements prepared in accordance with accounting principles generally accepted in the United States. We believe these condensed consolidated financial statements reflect all adjustments (consisting only of normal, recurring adjustments) that are necessary for a fair presentation of our financial position and results of operations for the periods presented. Results of operations for the interim periods presented are not necessarily indicative of results to be expected for the year. The information included in this Form 10-Q should be read in conjunction with the financial statements and notes thereto included in our fiscal 2005 Form 10-K on file with the SEC.
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent amounts in the financial statements and accompanying notes. Actual results could differ from those estimates. Certain prior year amounts have been reclassified to conform to the current year presentation. These reclassifications had no impact on net earnings as previously reported.
The unaudited condensed consolidated financial statements include the accounts ofAvanir Pharmaceuticals and its wholly-owned subsidiaries, Xenerex Biosciences and Avanir Holding Company. Intercompany accounts and transactions have been eliminated.
2. REVERSE STOCK SPLIT
On January 17, 2006, we implemented a one-for-four reverse stock split of our common stock. All share and per share information herein (including shares outstanding, earnings per share and warrant and stock option exercise prices) reflects this reverse split.
3. SIGNIFICANT ACCOUNTING POLICIES
Change in Accounting Method for Share-Based Compensation
We adopted the provisions of revised Statement of Financial Accounting Standards No. 123 (“FAS 123R”), “Share-Based Payment,” on October 1, 2005, the first day of our fiscal 2006, using the modified prospective transition method to account for our employee share-based awards. The valuation provisions of FAS 123R apply to new awards and to awards that are outstanding at the effective date and subsequently modified or cancelled. Estimated compensation expense for awards outstanding at the effective date will be recognized over the remaining service period using the compensation cost calculated for pro forma disclosure purposes under Statement of Financial Accounting Standards No. 123, “Accounting for Stock-Based Compensation” (“FAS 123”). Our consolidated financial statements as of March 31, 2006 and for the three-month and six-month periods ended March 31, 2006 reflect the impact of FAS 123R. In accordance with the modified prospective transition method, our consolidated financial statements for prior periods were not restated to reflect, and do not include, the impact of FAS 123R.
In March 2005, the Securities and Exchange Commission issued Staff Accounting Bulletin No. 107 (“SAB 107”) relating to FAS 123R. The Company has applied the provisions of SAB 107 in its adoption of FAS 123R.
7
On November 10, 2005, the FASB issued FASB Staff Position No. FAS 123R-3, “Transition Election Related to Accounting for Tax Effects of Share-Based Payment Awards” (“FAS 123R-3”). We have elected to adopt the alternative transition method provided in FAS 123R-3. The alternative transition method includes a simplified method to establish the beginning balance of the additional paid-in capital pool (“APIC pool”) related to the tax effects of employee share-based compensation, which is available to absorb tax deficiencies recognized subsequent to the adoption of FAS 123R.
Share-based compensation expense recognized during a period is based on the value of the portion of share-based payment awards that is ultimately expected to vest during the period. Share-based compensation expense recognized in our consolidated statement of operations for the three-month and six-month periods ended March 31, 2006 included compensation expense for share-based payment awards granted prior to, but not yet vested as of, September 30, 2005 based on the grant date fair value estimated in accordance with the pro forma provisions of FAS 123 and share-based payment awards granted subsequent to September 30, 2005 based on the grant date fair value estimated in accordance with FAS 123R. For share awards granted in fiscal 2006, expenses are amortized under the straight-line attribution method. For share awards granted prior to fiscal 2006, expenses are amortized under the straight-line single option method prescribed by FAS 123. As share-based compensation expense recognized in the consolidated statement of operations for the three-month and six-month periods ended March 31, 2006 is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. FAS 123R requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Pre-vesting forfeitures were estimated to be approximately 8% for both officers and directors and 13% for other employees in the six-month period ended March 31, 2006 based on our historical experience. In our pro forma information required under FAS 123 for the periods prior to fiscal 2006, we accounted for forfeitures as they occurred.
The adoption of FAS 123R resulted in incremental share-based compensation expense of $755,000 and $1.2 million in the three-month and six-month periods ended March 31, 2006, respectively. The incremental share-based compensation caused our net loss to increase by the same amounts and basic and diluted loss per share to increase by $0.02 per share and $0.04 per share in the three-month and six-month periods ended March 31, 2006, respectively. Total compensation expense related to all of our share-based awards, recognized under FAS 123R, for the three-month and six month periods ended March 31, 2006 was comprised of the following:
| | | | | | | | | |
| | | Three Months Ended | | | Six Months Ended | |
| | | March 31, 2006 | | | March 31, 2006 | |
| Research and development expense | | $ | 185,990 | | | $ | 262,337 | |
| Selling, general and administrative expense | | | 571,730 | | | | 957,396 | |
| | | | | | | |
| Share-based compensation expense before taxes | | | 757,720 | | | | 1,219,733 | |
| Related income tax benefits | | | — | | | | — | |
| | | | | | | |
| Share-based compensation expense | | $ | 757,720 | | | $ | 1,219,733 | |
| | | | | | | |
| Net share-based compensation expense per common Share — basic and diluted | | $ | (0.02 | ) | | $ | (0.04 | ) |
| | | | | | | |
| | | | | | | | | |
| Share-based compensation expense from: | | | | | | | | |
| Stock options | | $ | 400,356 | | | $ | 647,054 | |
| Restricted stock awards | | | 328,159 | | | | 543,474 | |
| Restricted stock units | | | 29,205 | | | | 29,205 | |
| | | | | | | |
| Total | | $ | 757,720 | | | $ | 1,219,733 | |
| | | | | | | |
Since we have a net operating loss carryforward as of March 31, 2006, no excess tax benefits for the tax deductions related to share-based awards were recognized in the consolidated statement of operations.
8
Additionally, no incremental tax benefits were recognized from stock options exercised in the six-month period ended March 31, 2006 that would have resulted in a reclassification to reduce net cash provided by operating activities with an offsetting increase in net cash provided by financing activities. Compensation expense relating to employee share-based awards was not recognized during the three-month and six-month periods ended March 31, 2005.
Through fiscal 2005, we accounted for share-based awards to employees using the intrinsic value method in accordance with Accounting Principles Board Opinion No. 25, “Accounting for Stock Issued to Employees” and related interpretations and provided the required pro forma disclosures of FAS 123. Under the intrinsic value method, no share-based compensation expense had been recognized in our consolidated statement of operations for share-based awards to employees, because the exercise price of our stock options granted to employees equaled the fair market value of the underlying stock at the date of grant.
The following table summarizes the pro forma effect on our net loss and per share data if we had applied the fair value recognition provisions of FAS 123 to share-based employee compensation for the three-month and six-month periods ended March 31, 2005.
| | | | | | | | | |
| | | Three Months | | | Six Months Ended | |
| | | Ended March 31, 2005 | | | March 31, 2005 | |
| Net loss, as reported | | $ | (14,050,947 | ) | | $ | (21,139,536 | ) |
| Add: Share-based employee compensation expense included in reported net loss | | | — | | | | — | |
| Deduct: Total share-based employee compensation expense determined under fair value based method for all awards expense determined under fair value based method for all awards | | | (453,794 | ) | | | (732,457 | ) |
| | | | | | | |
| Pro forma net loss | | $ | (14,504,741 | ) | | $ | (21,871,993 | ) |
| | | | | | | |
| Net loss per share: | | | | | | | | |
| Basic and diluted — as reported | | $ | (0.57 | ) | | $ | (0.87 | ) |
| Basic and diluted — pro forma | | $ | (0.59 | ) | | $ | (0.90 | ) |
For employee stock options granted during the six-month period ended March 31, 2005, we determined pro forma compensation expense under the provisions of FAS 123 using the Black-Scholes model and the following assumptions: (1) an expected volatility of 120%, (2) an expected term of 3.4 years, (3) a risk-free interest rate of 3.0% and (4) an expected dividend yield of 0%. The weighted average fair value of options granted during the six-month period ended March 31, 2005 was $9.84 per share.
We account for stock options granted to non-employees in accordance with Emerging Issues Task Force No. 96-18, “Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services,” (“EITF 96-18”). Under EITF 96-18, we determine the fair value of the stock options granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable.
Revenue Recognition
We allocate revenue to separate elements in multiple element arrangements based on the guidance in Emerging Issues Task Force No. 00-21 (“EITF 00-21”), “Accounting for Revenue Arrangements with Multiple Deliverables.” Revenues allocated to deliverables are recognized in accordance with SEC Staff Accounting Bulletin Topic 13 (“SAB Topic 13”), “Revenue Recognition” and revenue is recognized when four basic criteria of revenue recognition are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services rendered; (3) the fee is fixed or determinable; and (4) collectibility is reasonably assured.
9
Revenue Arrangements with Multiple Deliverables. We have revenue arrangements whereby we deliver to the customer multiple products and/or services (multiple deliverables). Such arrangements may include antibody generation services agreements, licensed compounds and intellectual property, and research and development services. In accordance with EITF 00-21, we analyze our multiple element arrangements to determine whether the elements can be separated and accounted for individually as separate units of accounting. The evaluation is performed at the inception of the arrangement and as each element is delivered. The delivered item(s) is (are) considered a separate unit of accounting if all of the following criteria are met: (1) the delivered item(s) has (have) value to the customer on a standalone basis; (2) there is objective and reliable evidence of the fair value of the undelivered item(s); and (3) if the arrangement includes a general right of return relative to the delivered item(s), delivery, or performance of the undelivered item(s) is (are) considered probable and substantially in our control.
License Arrangements. License arrangements may consist of non-refundable upfront license fees, data transfer fees, or research reimbursement payments and/or exclusive licensed rights to patented or patent pending compounds, technology access fees, various performance or sales milestones and future product royalty payments. Such arrangements may also include fees for specific research and development services. If the delivered elements do not have standalone value or if we do not have objective or reliable evidence of the fair value of the undelivered component, the amount of revenue allocable to the delivered element is deferred. Non-refundable, up-front license fees, data transfer fees, and research reimbursement payments that have standalone value that are not contingent on any future performance by us, and that do not rely on our continuing involvement including any proprietary technology under the arrangements, and for which collection is assured, are recognized as revenue upon the delivery of the data and/or licensed compounds and commencement of the license term. Such deliverables may include physical quantities of compounds, design of the compounds and structure-activity relationships, the conceptual framework and mechanism of action, and rights to the patents or patents pending for such compounds. We consider licensed rights or licensed technology to have standalone value to our customers if we or others have sold such rights or technology separately or our customers can sell such rights or technology separately without the need for our continuing involvement. We defer revenue if we have continuing performance obligations without which the technology, right, product or service conveyed in conjunction with the non-refundable fee has no utility to the licensee that is separate and independent of our performance under the other elements of the arrangement. In addition, if we have continuing involvement through research and development services that are required because our know-how and expertise related to the technology is proprietary to us, or can only be performed by us, then such up-front fees are deferred and recognized over the period of continuing involvement.
Payments related to substantive, performance-based milestones are recognized as revenue upon the achievement of the milestones as specified in the underlying agreements, when they represent the culmination of the earnings process. Revenue from research and development services is recognized during the period in which the services are performed and is based upon the number of FTE personnel working on the specific project at the agreed-upon rate. Reimbursements from collaborative partners for agreed upon direct costs including direct material and outsourced, or subcontracted, pre-clinical studies are recognized as revenues in the period the reimbursable expenses are incurred. Payments received in advance are recorded as deferred revenue until the research and development services are performed, costs are incurred, or a milestone is reached.
Royalty Revenues.We recognize royalty revenues from licensed products when earned in accordance with the terms of the license agreements. Net sales figures used for calculating royalties include deductions for costs of unsaleable returns, managed care chargebacks, cash discounts, freight and warehousing, and miscellaneous write-offs, which may vary over the course of the license agreement.
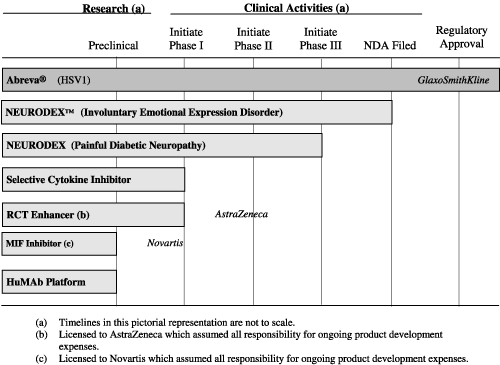
Revenues from Sale of Royalty Rights.In agreements where we have sold our rights to future royalties under license agreements and we maintain continuing involvement in earning such royalties, we defer revenues and recognize them over the life of the license agreement. For example, in the sale of an undivided interest of our Abreva® license agreement to Drug Royalty USA, revenue recognition is being determined under the “units-of-revenue method.” Under this method, the amount of deferred revenue to be recognized as revenue in each period is calculated by multiplying the following: (1) the ratio of the
10
royalty payments due to Drug Royalty USA for the period to the total remaining royalties that we expect GlaxoSmithKline will pay Drug Royalty USA over the term of the agreement by (2) the unamortized deferred revenue amount.
Government Research Grant Revenue.We recognize revenues from federal research grants during the period in which the related expenditures are incurred.
Product Sales — Active Pharmaceutical Ingredient Docosanol.Revenue from product sales, which consist of sales of our active pharmaceutical ingredient docosanol, is recorded when the above criteria are met, generally when title and risk of loss have passed to the buyer upon delivery. We sell the active pharmaceutical ingredient docosanol to various licensees and ship only on written order for the materials. Total shipments generally occur fewer than five times a year. Our contracts for sales of the active pharmaceutical ingredient docosanol include buyer acceptance provisions that give our buyers the right of replacement if the delivered materials do not meet specified criteria by giving notice within 30 days after receipt of such defective materials. We have the option to refund or replace any such defective materials however, we have historically demonstrated that the materials shipped from the same pre-inspected lot have consistently met the specified criteria and no buyer has rejected any of our shipments from the same pre-inspected lot to date. Therefore, we recognize revenue from sales of the active pharmaceutical ingredient docosanol without providing for such contingency upon shipment.
Recognition of Expenses in Outsourced Contracts
Pursuant to management’s assessment of the services that have been performed on clinical trials and other contracts, we recognize expenses as the services are provided. Such management assessments include, but are not limited to: (1) an evaluation by the project manager of the work that has been completed during the period, (2) measurement of progress prepared internally and/or provided by the third-party service provider, (3) analyses of data that justify the progress, and (4) management’s judgment.
Research and Development Expenses
Research and development expenses consist of expenses incurred in performing research and development activities including salaries and benefits, facilities and other overhead expenses, clinical trials, contract services and other outside expenses. Research and development expenses are charged to operations as they are incurred. Up-front payments to collaborators made in exchange for the avoidance of potential future milestone and royalty payments on licensed technology are also charged to research and development expense when the drug is still in the development stage, has not been approved by the FDA for commercialization and concurrently has no alternative uses.
We assess our obligations to make milestone payments that may become due under licensed or acquired technology to determine whether the payments should be expensed or capitalized. We charge milestone payments to research and development expense when:
| | • | | The technology is in the early stage of development and has no alternative uses; |
| |
| | • | | There is substantial uncertainty of the technology or product being successful; |
| |
| | • | | There will be difficulty in completing the remaining development; and |
| |
| | • | | There is substantial cost to complete the work. |
Acquired contractual rights. Payments to acquire contractual rights to a licensed technology or drug candidate are expensed as incurred when there is uncertainty in receiving future economic benefits from the acquired contractual rights. We consider the future economic benefits from the acquired contractual rights to a drug candidate to be uncertain until such drug candidate is approved by the FDA or when other significant risk factors are abated.
11
Capitalization and Valuation of Long-Lived and Intangible Assets
Intangible assets with finite useful lives consist of capitalized legal costs incurred in connection with patents, patent applications pending and license agreements. We amortize costs of approved patents, patent applications pending and license agreements over their estimated useful lives, or terms of the agreements, whichever are shorter. For patents pending, we amortize the costs over the shorter of a period of twenty years from the date of filing the application or, if licensed, the term of the license agreement. We re-assess the useful lives of patents when they are issued, or whenever events or changes in circumstances indicate the useful lives may have changed. For patent and patent applications pending and trademarks that we abandon, we charge the remaining unamortized accumulated costs to expense.
Intangible assets and long-lived assets are evaluated for impairment whenever events or changes in circumstances indicate that their carrying value may not be recoverable. If the review indicates that intangible assets or long-lived assets are not recoverable (i.e. the carrying amount is less than the future projected undiscounted cash flows), their carrying amount would be reduced to fair value. Factors we consider important that could trigger an impairment review include the following:
| | • | | A significant underperformance relative to expected historical or projected future operating results; |
| |
| | • | | A significant change in the manner of our use of the acquired asset or the strategy for our overall business; and/or |
| |
| | • | | A significant negative industry or economic trend. |
When we determine that the carrying value of intangible assets or long-lived assets are not recoverable based upon the existence of one or more of the above indicators of impairment, we may be required to record impairment charges for these assets.
Stock Offering Costs
Expenses incurred in connection with common stock issuances are recorded as an offset to common stock on the condensed consolidated balance sheets. These expenses consist of underwriters’ discounts and commissions and third-party related offering expenses.
4. INVESTMENTS
Investments in securities as of March 31, 2006 and September 30, 2005 consisted of the following:
| | | | | | | | | | | | | | | | | |
| | | Amortized | | | Gross Unrealized | | | Gross | | | Fair | |
| | | Cost | | | Gains (1) | | | Unrealized Losses (1) | | | Value | |
| As of March 31, 2006: | | | | | | | | | | | | | | | | |
| Certificates of deposit | | $ | 856,597 | | | $ | — | | | $ | — | | | $ | 856,597 | |
| Government debt securities | | | 41,490,224 | | | | — | | | | (305,145 | ) | | | 41,185,079 | |
| | | | | | | | | | | | | |
| Total | | $ | 42,346,821 | | | $ | — | | | $ | (305,145 | ) | | $ | 42,041,676 | |
| | | | | | | | | | | | | |
| Classified and reported as: | | | | | | | | | | | | | | | | |
| Short term investments: | | | | | | | | | | | | | | | | |
| Classified as available-for-sale | | | | | | | | | | | | | | $ | 38,738,504 | |
| | | | | | | | | | | | | | | | |
| Long term investments: | | | | | | | | | | | | | | | | |
| Classified as available-for-sale | | | | | | | | | | | | | | | 2,446,575 | |
| Restricted investments in securities (2) | | | | | | | | | | | | | | | 856,597 | |
| | | | | | | | | | | | | | | | |
| Long-term investments | | | | | | | | | | | | | | | 3,303,172 | |
| | | | | | | | | | | | | | | | |
| Total | | | | | | | | | | | | | | $ | 42,041,676 | |
| | | | | | | | | | | | | | | | |
12
| | | | | | | | | | | | | | | | | |
| | | | | | | Gross Unrealized | | | Gross Unrealized | | | Fair | |
| | | Amortized Cost | | | Gains (3) | | | Losses (3) | | | Value | |
| As of September 30, 2005: | | | | | | | | | | | | | | | | |
| Certificates of deposit | | $ | 956,872 | | | $ | 48 | | | $ | — | | | $ | 956,920 | |
| Government debt securities | | | 18,074,385 | | | | — | | | | (113,862 | ) | | | 17,960,523 | |
| | | | | | | | | | | | | |
| Total | | $ | 19,031,257 | | | $ | 48 | | | $ | (113,862 | ) | | $ | 18,917,443 | |
| | | | | | | | | | | | | |
| Classified and reported as: | | | | | | | | | | | | | | | | |
| Short term investments: | | | | | | | | | | | | | | | | |
| Classified as available-for-sale | | | | | | | | | | | | | | $ | 14,215,005 | |
| | | | | | | | | | | | | | | | |
| Long term investments: | | | | | | | | | | | | | | | | |
| Classified as available-for-sale | | | | | | | | | | | | | | | 3,845,566 | |
| Restricted investments in securities (2) | | | | | | | | | | | | | | | 856,872 | |
| | | | | | | | | | | | | | | | |
| Long-term investments | | | | | | | | | | | | | | | 4,702,438 | |
| | | | | | | | | | | | | | | | |
| Total | | | | | | | | | | | | | | $ | 18,917,443 | |
| | | | | | | | | | | | | | | | |
| (1) | | Gross unrealized losses of $305,145 on government debt securities are reported as “accumulated other comprehensive loss” on the consolidated balance sheet as of March 31, 2006. |
| (2) | | Represents amounts pledged to our bank as collateral for letters of credit issued in connection with our leases of office and laboratory space. |
| (3) | | Gross unrealized gains of $48 and gross unrealized losses of $113,862 on government securities and certificates of deposit represent an accumulated net unrealized loss of $113,814, which is reported as “accumulated other comprehensive loss” on the consolidated balance sheet as of September 30, 2005. |
5. RECEIVABLES, NET
Receivables as of March 31, 2006 and September 30, 2005 consist of the following:
| | | | | | | | | |
| | | March 31, 2006 | | | September 30, 2005 | |
| Unbilled receivables | | $ | 1,249,087 | | | $ | 1,010,902 | |
| Receivables | | | 261,686 | | | | 186,851 | |
| | | | | | | |
| | | | 1,510,773 | | | | 1,197,753 | |
| Allowance for doubtful accounts | | | (28,099 | ) | | | (28,099 | ) |
| | | | | | | |
| Receivables, net | | $ | 1,482,674 | | | $ | 1,169,654 | |
| | | | | | | |
6. INVENTORY
Inventory includes active pharmaceutical ingredients docosanol, dextromethorphan, and quinidine sulfate as of March 31, 2006 and docosanol only as of September 30, 2005 with the following carrying amounts as of those dates:
| | | | | | | | | |
| | | March 31, 2006 | | | September 30, 2005 | |
| Inventory | | $ | 793,883 | | | $ | 374,539 | |
| Less: current portion | | | 271,375 | | | | 27,115 | |
| | | | | | | |
| Long-term portion | | $ | 522,508 | | | $ | 347,424 | |
| | | | | | | |
13
7. PROPERTY AND EQUIPMENT
Property and equipment consist of the following:
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | March 31, 2006 | | | September 30, 2005 | |
| | | Gross Carrying | | | Accumulated | | | | | | | Gross Carrying | | | Accumulated | | | | | |
| | | Value | | | Depreciation | | | Net | | | Value | | | Depreciation | | | Net | |
| Research and development equipment | | $ | 3,976,257 | | | $ | (2,860,591 | ) | | $ | 1,115,666 | | | $ | 3,903,735 | | | $ | (2,567,843 | ) | | $ | 1,335,892 | |
| Computer equipment and related software | | | 1,273,804 | | | | (661,122 | ) | | | 612,682 | | | | 1,038,390 | | | | (565,137 | ) | | | 473,253 | |
| Leasehold improvements | | | 5,676,238 | | | | (1,996,935 | ) | | | 3,679,303 | | | | 5,583,177 | | | | (1,641,485 | ) | | | 3,941,692 | |
| Office equipment, furniture and fixtures | | | 791,294 | | | | (349,215 | ) | | | 442,079 | | | | 558,911 | | | | (305,221 | ) | | | 253,690 | |
| Manufacturing equipment | | | 261,719 | | | | — | | | | 261,719 | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | |
| Total property and equipment | | $ | 11,979,312 | | | $ | (5,867,863 | ) | | $ | 6,111,449 | | | $ | 11,084,213 | | | $ | (5,079,686 | ) | | $ | 6,004,527 | |
| | | | | | | | | | | | | | | | | | | |
Depreciation expense related to property and equipment for the three-month and six-month periods ended March 31, 2006 was approximately $412,000 and $823,000, respectively, as compared to $384,000 and $802,000 for the three-month and six-month periods ended March 31, 2005, respectively. Manufacturing equipment represents the tooling purchased for the manufacture of Neurodex™, which had not been placed in service as of March 31, 2006. The manufacturing equipment will be used by our drug manufacturing services provider. See Note 16, “New Agreement,” for further discussion.
8. INTANGIBLE ASSETS
At March 31, 2006 and September 30, 2005, the components of amortizable and unamortizable intangibles are as follows:
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | March 31, 2006 | | | September 30, 2005 | |
| | | Gross Carrying | | | Accumulated | | | | | | | Gross Carrying | | | Accumulated | | | | | |
| | | Value | | | Depreciation | | | Net | | | Value | | | Depreciation | | | Net | |
| Intangible assets with finite lives: | | | | | | | | | | | | | | | | | | | | | | | | |
| Patent applications pending (1) | | $ | 3,068,996 | | | $ | (406,157 | ) | | $ | 2,662,839 | | | $ | 3,029,127 | | | $ | (336,457 | ) | | $ | 2,692,670 | |
| Patents (1) | | | 1,658,021 | | | | (644,259 | ) | | | 1,013,762 | | | | 1,429,532 | | | | (506,144 | ) | | | 923,388 | |
| Licenses | | | 42,461 | | | | (19,032 | ) | | | 23,429 | | | | 42,461 | | | | (17,904 | ) | | | 24,557 | |
| | | | | | | | | | | | | | | | | | | |
| Total intangible assets with finite lives | | | 4,769,478 | | | | (1,069,448 | ) | | | 3,700,030 | | | | 4,501,120 | | | | (860,505 | ) | | | 3,640,615 | |
| Intangible assets with indefinite useful lives: | | | | | | | | | | | | | | | | | | | | | | | | |
| Trademarks (2) | | | 26,134 | | | | — | | | | 26,134 | | | | 24,471 | | | | — | | | | 24,471 | |
| | | | | | | | | | | | | | | | | | | |
| Total intangible assets | | $ | 4,795,612 | | | $ | (1,069,448 | ) | | $ | 3,726,164 | | | $ | 4,525,591 | | | $ | (860,505 | ) | | $ | 3,665,086 | |
| | | | | | | | | | | | | | | | | | | |
| | |
| (1) | | Patent applications pending include the net effect of $4,910 and $39,638 (net of accumulated amortization of $0 and $3,409) in intangible assets abandoned during the three-month and six-month periods ended March 31, 2006, respectively. Patent applications pending include the net effect of $2,944 and $173,272 (net of accumulated amortization of $68 and $18,389) in intangible assets abandoned during the three-month and six-month periods ended |
14
| | |
| | | March 31, 2005, respectively. We abandoned certain patents pending related to docosanol 10% cream in selected countries where we determined there are no viable markets. |
| |
| (2) | | Intangible assets with indefinite useful lives include the net effect of $4,693 in trademarks abandoned during the three-month and six-month periods ended March 31, 2005. |
Amortization expense related to amortizable intangible assets for the three-month and six-month periods ended March 31, 2006 was approximately $69,000 and $133,000, respectively, compared to $55,000 and $102,000 for the three-month and six-month periods ended March 31, 2005, respectively. Based solely on the amortizable intangible assets as of March 31, 2006, the estimated annual amortization expense of intangible assets for the fiscal years ending September 30 is shown in the following table. Actual amortization expense to be reported in future periods could differ from these estimates as a result of acquisitions, divestitures, and other relevant factors.
Amortization Expense
| | | | | |
| Fiscal year ending September 30: | | | | |
| 2006 (remaining six months) | | $ | 136,000 | |
| 2007 | | | 270,000 | |
| 2008 | | | 270,000 | |
| 2009 | | | 262,000 | |
| 2010 | | | 234,000 | |
| Thereafter | | | 2,528,000 | |
| | | | |
| Total | | $ | 3,700,000 | |
| | | | |
9. DEFERRED REVENUE
The following table sets forth as of March 31, 2006 the deferred revenue balances for our sale of future Abreva® royalty rights to Drug Royalty USA and other agreements. The portion of deferred revenue classified as a current liability represents the amount we expect to realize as revenue within the next 12 months.
| | | | | | | | | | | | | |
| | | Drug Royalty | | | | | | | | | |
| | | USA Agreement | | | Other Agreements | | | Total | |
| Deferred revenue as of September 30, 2005 | | $ | 19,049,877 | | | $ | 108,333 | | | $ | 19,158,210 | |
| Changes during the period: | | | | | | | | | | | | |
| Additions during period | | | — | | | | 860,216 | | | | 860,216 | |
| Recognized as revenue during period | | | (1,009,123 | ) | | | (9,342 | ) | | | (1,018,465 | ) |
| | | | | | | | | | |
| Deferred revenue as of March 31, 2006 | | $ | 18,040,754 | | | $ | 959,207 | | | $ | 18,999,961 | |
| | | | | | | | | | |
| Classified and reported as: | | | | | | | | | | | | |
| Current portion of deferred revenue | | $ | 1,998,638 | | | $ | 317,480 | | | $ | 2,316,118 | |
| Deferred revenue, net of current portion | | | 16,042,116 | | | | 641,727 | | | | 16,683,843 | |
| | | | | | | | | | |
| Total deferred revenue | | $ | 18,040,754 | | | $ | 959,207 | | | $ | 18,999,961 | |
| | | | | | | | | | |
Drug Royalty Agreement— In December 2002, we sold to Drug Royalty USA an undivided interest in our rights to receive future Abreva® royalties under the license agreement with GlaxoSmithKline for $24.1 million (the “Drug Royalty Agreement” and the “GlaxoSmithKline License Agreement”, respectively). Under the Drug Royalty Agreement, Drug Royalty USA has the right to receive royalties from GlaxoSmithKline on sales of Abreva until December 2013.
In accordance with Staff Accounting Bulletin No. 101, “Revenue Recognition in Financial Statements,” (“SAB 101”), revenues are recognized when earned, collection is reasonably assured and no additional performance of services is required. We classified the proceeds received from Drug Royalty USA as deferred revenue, to be recognized as revenue ratably over the life of the license agreement
15
consistent with SAB 101 because of our continuing involvement over the term of the Drug Royalty Agreement. Such continuing involvement includes overseeing the performance of GlaxoSmithKline and its compliance with the covenants in the GlaxoSmithKline License Agreement, monitoring patent infringement and adverse claims or litigation involving Abreva® and undertaking to find a new license partner in the event that GlaxoSmithKline terminates the agreement. The Drug Royalty Agreement contains both covenants (Section 8) and events of default (Section 10) that require such performance on our part. Therefore, nonperformance on our part could result in default of the arrangement, and could give rise to additional rights in favor of Drug Royalty USA under a separate security agreement with Drug Royalty USA, which could result in loss of our rights to share in future Abreva royalties if wholesale sales by GlaxoSmithKline exceed $62 million a year. Because of our continuing involvement, we recorded the net proceeds of the transaction as deferred revenue, to be recognized as revenue ratably over the life of the license agreement. Based on a review of our continuing involvement, we concluded that the sale proceeds did not meet any of the rebuttable presumptions in EITF 88-18 that would require classification of the proceeds as debt.
Docosanol License Agreement— In January 2006, we signed an exclusive license agreement (the “License Agreement”) with Kobayashi Pharmaceutical Co., Ltd. (“Kobayashi”), a Japanese corporation, to allow Kobayashi to market in Japan medical products that are curative of episodic outbreaks of herpes simplex or herpes labialis and that contain a therapeutic concentration of our docosanol 10% cream either as the sole active ingredient or in combination with any other ingredient, substance or compound (the “Products”).
The License Agreement automatically expires upon the latest to occur of (1) the tenth anniversary of the first commercial sale in Japan, (2) the last expiration date of any patent licensed under the License Agreement, or (3) the last date of expiration of the post marketing surveillance period in Japan. Pursuant to the terms of the License Agreement, we received a non-refundable know-how and data transfer fee (“License Fee”) of $860,000 in March 2006. In addition, we will be eligible to receive milestone payments of up to 450 million Japanese Yen (or up to approximately U.S. $3.8 million based on the exchange rate as of March 31, 2006), subject to achievement of certain milestones relating to the regulatory approval and commercialization of docosanol in Japan and patent and know-how royalties for sales of Products in Japan, if commercial sales commence.
Under the terms of the License Agreement, Kobayashi will be responsible for obtaining all necessary approvals for marketing, all sales and marketing activities and the manufacturing and distribution of the Products. Because the know-how and expertise related to the docosanol 10% cream are proprietary to us, we will be providing assistance to Kobayashi, upon their request, in completing additional required clinical studies and filing the new drug application (“NDA”) submission for the licensed product in Japan. As of March 31, 2006, we estimated the period of time of our continuing involvement in advising and assisting Kobayashi with additional clinical studies and obtaining regulatory approval in Japan is three years. In accordance with SAB Topic 13, revenue from the License Fee of $860,000 is deferred and being recognized over three years. We recognized $9,000 of the License Fee in the three-month and six-month periods ended March 31, 2006.
10. COMPUTATION OF NET LOSS PER COMMON SHARE
Basic net (loss) earnings per common share is computed by dividing net loss by the weighted-average number of common shares outstanding during the period, excluding restricted stock that has been issued but is not yet vested. Diluted net (loss) earnings per common share is computed by dividing net loss by the weighted-average number of common shares outstanding during the period plus additional weighted average common equivalent shares outstanding during the period. Common equivalent shares result from the assumed exercise of outstanding stock options and warrants (the proceeds of which are then presumed to have been used to repurchase outstanding stock using the treasury stock method) and the vesting of unvested restricted shares of common stock. In the loss periods, the common equivalent shares have been excluded from the computation of diluted net loss per share, because their effect would have been anti-dilutive. For the three-month and six-month periods ended March 31, 2006 and 2005, 1,563,045 and 1,503,244 shares of common stock, respectively, issuable under our share-based compensation plans were excluded from the computation of diluted net loss per share. For the three-month and six-month periods ended March 31, 2006 and 2005, warrants to purchase 269,305 and 1,311,315 shares of common stock, respectively, were excluded from the computation of diluted net loss per share.
16
11. COMPREHENSIVE LOSS
Comprehensive loss consists of the following:
| | | | | | | | | | | | | | | | | |
| | | Three Months Ended March 31, | | | Six Months Ended March 31, | |
| | | 2006 | | | 2005 | | | 2006 | | | 2005 | |
| Net loss | | $ | (13,405,411 | ) | | $ | (14,050,947 | ) | | $ | (19,079,845 | ) | | $ | (21,139,536 | ) |
| Other comprehensive loss, net of tax: | | | | | | | | | | | | | | | | |
| Unrealized loss on available-for-sale securities method for all awards | | | (131,994 | ) | | | (41,464 | ) | | | (191,331 | ) | | | (529 | ) |
| | | | | | | | | | | | | |
| Total comprehensive loss | | $ | (13,537,405 | ) | | $ | (14,092,411 | ) | | $ | (19,271,176 | ) | | $ | (21,140,065 | ) |
| | | | | | | | | | | | | |
12. SHAREHOLDERS’ EQUITY
Unearned compensation. Pursuant to FAS 123R, unearned compensation with the balance of $3.5 million at September 30, 2005 was eliminated against common stock upon the adoption of FAS 123R on October 1, 2005. The unearned compensation was related to a restricted stock award granted to our Chief Executive Officer prior to the adoption of FAS 123R. See Note 3, “Significant Accounting Policies — Change in Accounting Method for Share-based Compensation.”
Common stock. In October 2005, we issued and sold to certain institutional investors 1,523,585 shares of our common stock at a price of $10.60 per share, for aggregate net offering proceeds of approximately $16.15 million. In December 2005, we issued and sold to certain institutional investors 1,492,538 shares of our common stock at $13.40 per share, for aggregate offering proceeds of approximately $20.0 million and net offering proceeds of approximately $19.4 million, after deducting commissions and offering fees and expenses. These offerings were made pursuant to our shelf registration statement on Form S-3, filed with the SEC in June 2005.
Between January 26, 2006 and February 7, 2006, we received proceeds of $4.7 million from the exercise of warrants to purchase 671,923 shares of Class A common stock in connection with our call for redemption of a group of outstanding warrants. The warrants had been issued in connection with a financing transaction in December 2003 involving the sale of Class A common stock and warrants (the “Warrants”). The exercise price of the Warrants was $7.00 per share. The Warrants had a five-year term, but included a provision that we could redeem the Warrants for $1.00 each if our stock price traded above twice the warrant exercise price for a certain period of time (the “Redemption Right”). On January 24, 2006, we sent the Warrant holders notice that the Redemption Right had been triggered and that the Warrants would expire, to the extent unexercised, on February 7, 2006. One of the warrants to purchase 25,167 shares of Class A common stock expired unexercised.
During the three-month period ended March 31, 2006, we issued an aggregate of 1,083,743 shares of common stock in connection with the exercises of Class A stock purchase warrants (673,936 shares at a weighted average exercise price of $7.01), employee stock options (397,307 shares at a weighted average exercise price of $6.66), and the exercise of an employee restricted stock award (12,500 shares at the purchase price of $0.001) for cash in the aggregate amount of $7.4 million.
As of March 31, 2006 and 2005, warrants to purchase 269,305 and 1,311,315 shares of common stock, respectively, at a weighted-average price per share of $8.92 and $7.27, respectively, remained outstanding, all of which are exercisable. As of March 31, 2006 and 2005, options to purchase 1,494,065 and 1,503,244 shares of common stock, respectively, at a weighted-average price per share of $10.62 and $8.54, respectively, remained outstanding; of these, 877,031 and 1,198,298 options were exercisable at March 31, 2006 and 2005, respectively. As of March 31, 2006 and 2005, 68,980 and 0 shares of common stock, respectively, are reserved for issuance under agreements to issue shares in connection with restricted stock awards and restricted stock units (“RSUs”).
17
13. EQUITY INCENTIVE PLANS
We currently have five equity incentive plans (the “Plans”): the 2005 Equity Incentive Plan, the 2003 Equity Incentive Plan, the 2000 Stock Option Plan, the 1998 Stock Option Plan and the 1994 Stock Option Plan. All of the Plans were approved by the shareholders, except for the 2003 Equity Incentive Plan, which was approved solely by the Board of Directors. Stock-based awards are subject to terms and conditions established by the Compensation Committee of our Board of Directors. Our policy is to issue new common shares upon the exercise of stock options, conversion of share units or purchase of restricted stock.
During the three-month and six-month periods ended March 31, 2006, share-based awards were granted under the 2003 Equity Incentive Plan (the “2003 Plan”) and the 2005 Equity Incentive Plan (the “2005 Plan”). Under the 2003 Plan and 2005 Plan, options to purchase shares, restricted stock units, restricted stock and other share-based awards may be granted to our employees and consultants. Under the Plans, as of March 31, 2006, we had an aggregate of 2,303,243 shares of our common stock reserved for issuance. Of those shares, 1,563,045 were subject to outstanding options and other awards and 740,198 shares were available for future grants of share-based awards. None of the share-based awards is classified as a liability as of March 31, 2006.
Stock Options. Stock options are generally granted with an exercise price equal to the current market price of our common stock at the grant date and have 10-year contractual terms. Options awards typically vest in accordance with one of the following schedules:
| | a. | | 25% of the option shares vest and become exercisable on the first anniversary of the grant date and the remaining 75% of the option shares vest and become exercisable quarterly in equal installments thereafter over three years; |
| |
| | b. | | One-third of the option shares vest and become exercisable on the first anniversary of the grant date and the remaining two-thirds of the option shares vest and become exercisable daily or quarterly in equal installments thereafter over two years; or |
| |
| | c. | | Options fully vest and become exercisable at the date of grant. |
Certain option awards provide for accelerated vesting if there is a change in control (as defined in the Plans).
Summaries of stock options outstanding and changes under the Plans during the six-month period ended March 31, 2006 are presented below.
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | Weighted average | | | | |
| | | | | | | Weighted average | | | remaining | | | Aggregate intrinsic | |
| | | | | | | exercise price per | | | contractual term | | | value | |
| | | Number of shares | | | share | | | (in years) | | | (in millions) | |
| Outstanding, September 30, 2005 | | | 1,600,034 | | | $ | 8.76 | | | | | | | | | |
| Granted | | | 361,812 | | | $ | 11.00 | | | | | | | | | |
| Exercised | | | (455,304 | ) | | $ | 6.23 | | | | | | | | | |
| Forfeited or expired expense determined under fair value based method for all awards | | | (12,477 | ) | | $ | 11.94 | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| Outstanding, March 31, 2006 | | | 1,494,065 | | | $ | 10.62 | | | | 7.0 | | | $ | 6.4 | |
| | | | | | | | | | | | | | | | |
| |
| Exercisable, March 31, 2006 | | | 877,031 | | | $ | 9.02 | | | | 5.2 | | | $ | 5.1 | |
| | | | | | | | | | | | | | | | |
18
The weighted average grant-date fair values of options granted during the six-month periods ended March 31, 2006 and 2005 were $8.57 per share and $9.84 per share, respectively. The total intrinsic value of options exercised during the six-month periods ended March 31, 2006 and 2005 was $4.3 million and $144,000, respectively, based on the differences in market prices on the dates of exercise and the option exercise prices.
The fair value of each option award is estimated on the date of grant using the Black-Scholes-Merton option pricing model (“Black-Scholes model”) that uses the assumptions noted in the following table. Expected volatilities are based on historical volatility of our common stock and other factors. The expected term of options granted is based on analyses of historical employee termination rates and option exercises. The risk-free interest rates are based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. Assumptions used in the Black-Scholes model for options granted during the six-month period ended March 31, 2006 were as follows:
| | | |
| Expected volatility | | 78.4%-80.4% |
| Weighted-average volatility | | 79.0% |
| Average expected term in years | | 4.5 |
| Risk-fee interest rate (zero coupon U.S. Treasury Note) | | 4.4% |
| Expected dividend yield | | 0% |
Restricted stock units.RSUs generally vest based on three years of continuous service and may not be sold or transferred until the awardee’s termination of service. The following table summarizes the RSU activities for the six-month period ended March 31, 2006:
| | | | | | | | | |
| | | | | | | Weighted average | |
| | | | | | | grant date | |
| | | Number of shares | | | fair value | |
| Nonvested, September 30, 2005 | | | — | | | $ | — | |
| Granted | | | 51,480 | | | $ | 15.54 | |
| | | | | | | | |
| Nonvested, March 31, 2006 | | | 51,480 | | | $ | 15.54 | |
| | | | | | | | |
The grant-date fair value of RSUs granted during the six-month period ended March 31, 2006 was $800,000. No RSUs were granted during the six-month period ended March 3, 2005. As of March 31, 2006, the total unrecognized compensation cost related to unvested shares was $667,000, which is expected to be recognized over a weighted-average period of 2.9 years, based on the vesting schedules.
Restricted stock awards. Restricted stock awards are grants that entitle the holder to acquire shares of restricted common stock at a fixed price, which is typically nominal. The shares of restricted stock cannot be sold, pledged, or otherwise disposed of until the award vests and any unvested shares may be reacquired by us for the original purchase price following the awardee’s termination of service. The restricted stock awards typically vest on the second or third anniversary of the grant date or on a graded vesting schedule over three years of employment. A summary of our nonvested restricted stock awards as of March 31, 2006 and changes during the six-month period then ended are presented below.
| | | | | | | | | |
| | | | | | | Weighted average | |
| | | | | | | grant date fair | |
| | | Number of shares | | | value | |
| Nonvested, September 30, 2005 | | | 250,000 | | | $ | 14.22 | |
| Granted | | | 30,000 | | | $ | 15.12 | |
| | | | | | | | |
| Nonvested, March 31, 2006 | | | 280,000 | | | $ | 14.32 | |
| | | | | | | �� | |
19
The grant-date fair value of restricted stock awards granted during the six-month period ended March 31, 2006 was $454,000. No restricted stock awards were granted in the six-month period ended March 31, 2005. As of March 31, 2006, the total unrecognized compensation cost related to unvested shares was $2.8 million, which is expected to be recognized over a weighted-average period of 2.4 years.
For the six-month period ended March 31, 2006, we received a total of $2.8 million in cash from options exercised under all share-based payment arrangements.
14. COMMITMENTS AND CONTINGENCIES
In the ordinary course of business, we may face various claims brought by third parties, including claims relating to employment and the safety or efficacy of our products. Any of these claims could subject us to costly litigation and, while we generally believe that we have adequate insurance to cover many different types of liabilities, our insurance carriers may deny coverage or our policy limits may be inadequate to fully satisfy any damage awards or settlements. If this were to happen, the payment of any such awards could have a material adverse effect on our operations, cash flows and financial position. Additionally, any such claims, whether or not successful, could damage our reputation and business. Management believes the outcome of currently identified claims and lawsuits will not likely have a material adverse effect on our operations or financial position.
15. LICENSE AGREEMENTS
AstraZeneca UK Limited (“AstraZeneca”). In July 2005, we entered into an exclusive license and research collaboration agreement with AstraZeneca to discover, develop and commercialize reverse cholesterol transport enhancing compounds for the treatment of cardiovascular disease. Under the terms of the agreement, we will be eligible to receive royalty payments, assuming the product is successfully developed by AstraZeneca and approved for marketing by the FDA. We are also eligible to receive up to $330 million in milestone payments contingent upon achievement of certain development and regulatory milestones by AstraZeneca, which could take several years of further development, including achievement of certain sales targets, if a licensed compound is approved for marketing by the FDA. Under this agreement, we received a license fee of $10 million in July 2005 and will also receive research funding of between $2.5 million to $4.0 million per year for providing research and development services
20
to AstraZeneca for up to three years. AstraZeneca assumed responsibility for the development and commercialization of the product.
Novartis International Pharmaceutical Ltd. (“Novartis”). In April 2005, we entered into a research, development and commercialization agreement with Novartis for orally active small molecule therapeutics that regulate macrophage migration inhibitory factor (“MIF”) in the treatment of various inflammatory diseases. Under the terms of the agreement, we will be eligible to receive royalty payments, assuming the product is successfully developed and approved by Novartis for marketing by the FDA. We are also eligible to receive up to $198 million in milestone payments contingent upon achievement of certain development and regulatory milestones by Novartis, including approval for certain additional indications, and regulatory milestones, which could take several years of further development by Novartis, including achievement of certain sales targets, when and if a MIF compound is approved for marketing by the FDA. Additionally, we received a license fee of $2.5 million in May 2005 for the license fee and transfer of data and know-how of the MIF technology to Novartis. We will also receive research funding of between $1.5 million and $2.5 million per year for providing research and development services to Novartis for two years from the date of the agreement, or longer upon mutual agreement of the parties. Novartis assumed responsibility for all development expenses.
Payments related to substantive, performance-based milestones are recognized as revenue upon the achievement of the milestones as specified in the agreement. Revenue from research and development services is recognized during the period in which the services are performed and is based upon the number of FTE personnel working on the project at the agreed-upon rates. During the three-month and six months ended March 31, 2006, we recognized revenues in connection with the collaborative agreements with AstraZeneca and Novartis as follows:
| | | | | | | | | |
| | | Three Months | | | Six Months Ended | |
| | | Ended March 31, 2006 | | | March 31, 2006 | |
Revenues: | | | | | | | | |
| Milestone revenue | | $ | — | | | $ | 5,000,000 | |
| Research services and reimbursements | | | 1,956,565 | | | | 4,434,583 | |
| | | | | | | |
| Total | | $ | 1,956,565 | | | $ | 9,434,583 | |
| | | | | | | |
16. MANUFACTURING SERVICES AGREEMENT
In January 2006, we entered into a manufacturing services agreement with Patheon Inc. (“Patheon”), a provider of outsourced drug manufacturing services, under which Patheon will initially serve as our sole provider of drug manufacturing services for Neurodex™ (the “Manufacturing Agreement”). Under the terms of the Manufacturing Agreement, Patheon will be responsible for manufacturing, quality control, quality assurance and stability testing of Neurodex. The Manufacturing Agreement is effective until December 31, 2011.
17. SEGMENT INFORMATION
We operate our business on the basis of a single reportable segment, which is the business of discovery, development and commercialization of novel therapeutics for chronic diseases. Our chief operating decision-maker is the Chief Executive Officer, who reviews our operating results on an aggregate basis and manages our operations as a single operating segment. We have developed one commercial product, docosanol 10% cream, known as Abreva® in North America, and we have several other product candidates in various stages of development. We have licensed docosanol 10% cream to other companies in the world that market the product and provide us royalties on product sales. We also have exclusively licensed to AstraZeneca compounds for the potential treatment of atherosclerosis and we provide them with research services. We also have exclusively licensed to Novartis compounds that target macrophage migration inhibitory factor. We earn license fee and research service revenues from AstraZeneca and
21
Novartis. These collaborative agreements with AstraZeneca and Novartis will also provide: (1) milestone payments if certain development and regulatory milestones are achieved, which could take several years of further development, (2) milestone payments upon achievement of certain sales targets, if the products are approved for marketing by the FDA, and (3) royalties on sales, if the products are approved for marketing by the FDA.
We categorize revenues by geographic area based on selling location. All our operations are currently located in the United States; therefore, total revenues for the three-month and six-month periods ended March 31, 2006 and 2005 are attributed to the United States. All long-lived assets at March 31, 2006 and September 30, 2005 are located in the United States.
Revenues derived from our license agreement with AstraZeneca for the three-month and six-month periods ended March 31, 2006 were 59% and 79%, respectively, of our total consolidated revenues. Revenues from our license agreement with Novartis for the three-month and six-month periods ended March 31, 2006 were 20% and 9%, respectively, of our total consolidated revenues. Revenues from the sale of rights to royalties under the GlaxoSmithKline license agreement for the three-month periods ended March 31, 2006 and 2005 were 17% and 65%, respectively, of our total consolidated revenues and 10% and 61% of our total consolidated revenues for the six-month periods ended March 31, 2006 and 2005, respectively. As of March 31, 2006 and September 30, 2005, receivables from AstraZeneca accounted for approximately 74% and 81%, respectively, of our total net receivables. As of March 31, 2006 and September 30, 2005, receivables from Novartis accounted for approximately 10% and 5%, respectively, of our total net receivables.
18. RECENT ACCOUNTING PRONOUNCEMENTS
Financial Accounting Standards No. 151 (“FAS 151”).In November 2004, the FASB issued FAS 151, “Inventory Costs”, which requires abnormal amounts of idle facility expense, freight, handling costs and wasted material (spoilage), as well as unallocated overhead, to be recognized as current period charges. FAS 151 became effective for inventory costs incurred beginning October 1, 2005. Adoption of FAS 151 did not significantly affect our financial condition or results of operations.
Financial Accounting Standards No. 154 (“FAS 154”).In May 2005, the FASB issued FAS 154, “Accounting Changes and Error Corrections.” FAS 154 establishes retrospective application as the required method for reporting a change in accounting principle in the absence of explicit transition requirements specific to the newly adopted accounting principle. FAS 154 also provides guidance for determining whether retrospective application of a change in accounting principle is impracticable and for reporting a change when retrospective application is impracticable. FAS 154 is effective for accounting changes and corrections of errors made in fiscal years beginning after December 15, 2005. We do not expect the adoption of FAS 154 to significantly affect our financial condition or results of operations.
Financial Accounting Standards No. 155 (“FAS 155”).In February 2006, the FASB issued FAS 155, “Accounting for Certain Hybrid Financial Instruments,” an amendment of Financial Accounting Standards No. 133, “Accounting for Derivative Instruments and Hedging Activities” (“FAS 133”) and Financial Accounting Standards No. 140, “Accounting for Transfers and Servicing of Financial Assets and Extinguishments of Liabilities” (“FAS 140”). With respect to FAS 133, FAS 155 simplifies accounting for certain hybrid financial instruments by permitting fair value remeasurement for any hybrid financial instrument that contains embedded derivative that otherwise would require bifurcation and eliminates the interim guidance in Statement 133 Implementation Issue No. D1, “Application of Statement 133 to Beneficial Interests in Securitized Financial Assets,” which provided that beneficial interests in securitized financial assets are not subject to the provision of FAS 133. With respect to FAS 140, FAS 155 eliminates a restriction on the passive derivative instruments that a qualifying special-purpose entity may hold. FAS 155 is effective for all financial instruments acquired or issued after the beginning of an entity’s first fiscal year that begins after September 15, 2006. We do not expect the adoption of FAS 155 to significantly affect our financial condition or results of operations.
22
19. SUBSEQUENT EVENTS
On May 4, 2006 our Board of Directors authorized a space restructuring plan and the relocation of our commercial and general and administrative operations. We currently occupy three laboratory buildings in San Diego, California, and plan on relocating all operations other than research and development to Orange County and subleasing two of the buildings in San Diego after the relocation. On May 5, 2006, we entered into a lease for the new Orange County facilities, which will consist initially of approximately 11,000 square feet of office space, increasing to 17,000 square feet in the second quarter of 2007, leased for an initial term of five years. The aggregate minimum payments over the initial term of the lease are expected to be approximately $2.8 million.
23
Item 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This Quarterly Report on Form 10-Q contains forward-looking statements concerning future events and performance of the Company. When used in this report, the words “intend,” “estimate,” “anticipate,” “believe,” “plan” or “expect” and similar expressions are included to identify forward-looking statements. These forward-looking statements are based on our current expectations and assumptions and many factors could cause our actual results to differ materially from those indicated in these forward-looking statements. You should review carefully the factors identified in this report under the caption, “Risk Factors” and in our most recent Annual Report on Form 10-K filed with the Securities and Exchange Commission (“SEC”). We disclaim any obligation to update or announce revisions to any forward-looking statements to reflect actual events or developments. Except as otherwise indicated herein, all dates referred to in this report represent periods or dates fixed with reference to the calendar year, rather than our fiscal year ending September 30. The three-month and six-month periods ended March 31, 2006 may also be referred to as the second quarter of fiscal 2006 and first half of fiscal 2006, respectively.
EXECUTIVE OVERVIEW
Avanir Pharmaceuticals is focused on developing and commercializing novel therapeutic products for the treatment of chronic diseases. Our product candidates address therapeutic markets including those for the central nervous system, atherosclerosis, inflammatory diseases and infectious diseases. Our research and drug discovery programs are focused primarily on small molecules that can be taken orally as therapeutic treatments.
Our recent developments are as follows:
| | • | | On April 4, 2006, we announced that the U.S. Federal Drug and Administration (“FDA”) had accepted for filing and review our new drug application (“NDA”) for Neurodex™ for the treatment of Involuntary Emotional Expression Disorder (“IEED”), also known as pseudobulbar affect (“PBA”) or emotional lability. We were also informed by the FDA that the NDA would be granted priority review status. The FDA grants priority review status to products that, if approved, would address an unmet medical need or are considered to be potentially significant therapeutic advancements over existing approved therapies in the treatment, diagnosis or prevention of a disease. We completed the submission of the NDA on January 27, 2006 and we expect the FDA will take action on the NDA by July 30, 2006 (the “PDUFA date”), based on our estimates of the timing for the priority review. However, the action may be delayed if there are questions from the FDA about our NDA. |
| |
| | • | | On April 3, 2006, the NASDAQ® approved our application for listing our common stock for trading on the NASDAQ National Market System. On April 11, 2006, our stock began trading on the NASDAQ under the stock symbol “AVNR” simultaneously with the discontinuation of trading on the American Stock Exchange. |
| |
| | • | | On January 17, 2006, we implemented a one-for-four reverse stock split of our common stock. Authorization to implement the reverse stock split had been previously approved on March 17, 2005 by our shareholders at our annual meeting of shareholders. Our common stock began trading on the American Stock Exchange on a split-adjusted basis on January 18, 2006, and temporarily had a ticker symbol of “AVN.R” from January 18, 2006 through April 10, 2006. All share and per share information herein (including shares outstanding, earnings per share and warrant and stock option exercise prices) reflect the adjustment of the reverse stock split. |
| |
| | • | | In January 2006, we signed an exclusive license agreement with Kobayashi Pharmaceutical Co., Ltd. (“Kobayashi”) giving Kobayashi the rights to manufacture and sell docosanol 10% cream as a treatment for cold sores in Japan. |
24
| | • | | In January 2006, we entered into a manufacturing services agreement with Patheon, Inc. (“Patheon”) under which Patheon will initially serve as our sole provider of drug manufacturing services for Neurodex™ (the “Manufacturing Agreement”). Under the terms of the Manufacturing Agreement, Patheon will be responsible for manufacturing, quality control, quality assurance and stability testing of Neurodex. The Manufacturing Agreement is effective until December 31, 2011. |
| |
| | • | | During the quarter ended March 31, 2006, we continued to fill important new management positions, including the two positions of Vice President of Medical Affairs and Vice President of Sales. |
| |
| | • | | On May 4, 2006 our Board of Directors authorized a space restructuring plan and the relocation of our commercial and general and administrative operations. We currently occupy three laboratory buildings in San Diego, California, and plan on relocating all operations other than research and development to Orange County and subleasing two of the buildings in San Diego after the relocation. On May 5, 2006, we entered into a lease for the new Orange County facilities, which will consist initially of approximately 11,000 square feet of office space, increasing to 17,000 square feet in the second quarter of 2007, leased for an initial term of five years. The aggregate minimum payments over the initial term of the lease are expected to be approximately $2.8 million. |
| |
| | • | | On May 4, 2006, Michael J. Puntoriero joined us as Senior Vice President of Finance and, effective May 15, 2006, will be appointed Chief Financial Officer. As a key member of the senior executive team, Mr. Puntoriero will be responsible for the areas of finance, treasury, business planning, investor relations, information technology, and corporate development. Effective May 15, 2006, Gregory P. Hanson, who is currently our Vice President and Chief Financial Officer, will assume the new position of Vice President and Chief Accounting Officer. |
We are currently developing Neurodex for the treatment of IEED/PBA, and for the treatment of painful diabetic neuropathy. IEED/PBA is a complex neurological syndrome that is characterized by a lack of control of emotional expression, typically episodes of involuntary or exaggerated motor expression of emotion such as laughing and/or crying or weeping when the patient does not feel those emotions or in an exaggerated amount. IEED/PBA afflicts patients with neurological disorders such as amyotrophic lateral sclerosis (“ALS”), Alzheimer’s disease (“AD”), multiple sclerosis (“MS”), stroke, traumatic brain injury and Parkinson’s disease. While the exact number is unknown, the medical literature indicates that there are approximately 800,000 to 1,000,000 patients who have IEED/PBA in North America. If the FDA approves Neurodex, it would be the first drug approved for the treatment of IEED/PBA. Additionally, we are currently engaged in a Phase III
25
clinical trial with Neurodex in patients with painful diabetic neuropathy and we are evaluating Neurodex for use for other clinical indications.
We have two programs in Phase I clinical development. One development program is for the treatment of atherosclerosis and is partnered with AstraZeneca. In the second half of December 2005, we successfully submitted an investigational new drug application (“IND”) to the FDA for a compound under development, AZD2479 (also known as AVP-26452), that acts as a reverse cholesterol transport enhancer. AstraZeneca paid us $5.0 million in connection with the achievement of this milestone under the agreement. Our other development program is for AVP13358 (a selective cytokine inhibitor) and various backup compounds that are currently being evaluated in the treatment of systemic lupus erythematosus (“SLE”) .
Our pre-clinical research program targeting macrophage migration inhibitory factor (“MIF”) in the treatment of inflammatory diseases is partnered with Novartis International Pharmaceutical Ltd. (“Novartis”). Using our proprietary Xenerex™ technology, we continue to conduct research to develop injectable human monoclonal antibody products for anthrax and other infectious diseases.
Our first commercialized product, docosanol 10% cream, (sold as Abreva® by our marketing partner GlaxoSmithKline Consumer Healthcare in North America) is the only over-the-counter treatment for cold sores that has been approved by the FDA.
The following chart illustrates the status of research activities for our products, product candidates and licensed technologies that are commercialized or under development as of April 30, 2006.
We have historically sought to maintain flexibility in our cost structure by actively managing outsourced functions, such as clinical trials, legal counsel, documentation and testing of internal controls, pre-clinical development work, and manufacturing, warehousing and distribution services, rather than maintaining all of these functions in house. We believe the benefits of outsourcing, being flexible and being able to
26
rapidly respond to program delays or successes far outweigh the higher costs often associated with outsourcing at this stage of our development, although we expect more of these functions will be brought in-house as we grow and prepare for the potential commercial launch of Neurodex.
If the Neurodex NDA is approved by the FDA, we intend to begin marketing and selling the product within several months of receiving product approval. We are in the process of transforming from a research and development organization into a vertically integrated pharmaceutical company. In order to facilitate that transformation, we are continuing to build the necessary infrastructure to support the potential commercial launch of Neurodex. We are in the process of developing our sales and marketing strategy and are continuing to recruit sales and marketing personnel for key positions within the organization.
We intend to continue to seek partnerships with pharmaceutical companies to help fund research and development programs in exchange for sharing in the rights to commercialize new drugs. We may also seek to develop new drug candidates through research collaborations with other pharmaceutical companies, allowing us to share the risks and the opportunities that come from such development efforts. Additionally, we anticipate acquiring other drugs to leverage the infrastructure and sales organization being established to market Neurodex once it is approved for IEED. We expect that our selling, marketing, development and other operational costs will continue to exceed revenues from existing sources through at least fiscal 2007. Trends in revenues and various types of expenses are discussed further in the “Results of Operations.” We potentially will have to raise additional capital to prepare for and execute a product launch of Neurodex™ for IEED/PBA, if approved by the FDA for marketing, and to fund our ongoing clinical trials for Neurodex for painful diabetic neuropathy, as well as selected research and other operating activities. Our future capital needs will depend substantially on our ability to reach predetermined milestones under our existing collaboration agreements, as well as the economic terms and the timing of any new partnerships or collaborative arrangements with pharmaceutical companies under which we would expect our partners to fund the costs of such activities. If we are unable to raise capital as needed to fund our operations, or if we are unable to reach these milestones or enter into any such collaborative arrangements, then we may need to slow the rate of development of some of our programs or sell the rights to one or more of our drug candidates, and our commercialization plans for Neurodex may be adversely affected. For additional information about the risks and uncertainties that may affect our business and prospects, please see “Risk Factors.”
Our address of record is 11388 Sorrento Valley Road, San Diego, California 92121. Our telephone number is (858) 622-5200 and our e-mail address isinfo@avanir.com. Additional information aboutAvanir can be found on our website, atwww.avanir.com, and in our periodic and current reports filed with the SEC. Copies of our current and periodic reports filed with the SEC are available at the SEC Public Reference Room at 450 Fifth Street, N.W., Washington, D.C. 20549, and online atwww.sec.govand our website.No portion of our website is incorporated by reference into this report.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
Our discussion and analysis of our financial condition and results of operations is based upon our condensed consolidated financial statements prepared in accordance with generally accepted accounting principles in the United States. The preparation of these financial statements requires us to make a number of assumptions and estimates that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reported periods. These items are monitored and analyzed by management for changes in facts and circumstances, and material changes in these estimates could occur in the future. Changes in estimates are recorded in the period in which they become known. We base our estimates on historical experience and various other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from our estimates if past experience or other assumptions do not turn out to be substantially accurate.
27
We believe that the application of our accounting policies for share-based compensation expense, revenue recognition, expenses in outsourced contracts, research and development expenses and valuation of long-lived and intangible assets, all of which are important to our financial position and results of operations, require significant judgments and estimates on the part of management.
Share-based compensation expense
We grant options to purchase our common stock to our employees, directors and consultants under our stock option plans. The benefits provided under these plans are share-based payments subject to the provisions of revised Statement of Financial Accounting Standards No. 123, “Share-Based Payment” (“FAS 123R”). Effective October 1, 2005, we adopted FAS 123R and use the fair value method to account for share-based payments with a modified prospective application which provides for certain changes to the method for valuing share-based compensation. The valuation provisions of FAS 123R apply to new awards and to awards that are outstanding on the effective date and subsequently modified or cancelled. Under the modified prospective application, prior periods are not revised for comparative purposes. Total compensation costs for our share-based payments recognized in the three-month and six-month periods ended March 31, 2006 was $758,000 and $1.2 million, respectively. Selling, general and administrative expense in the three-month and six-month periods ended March 31, 2006 included share-based compensation of $572,000 and $957,000, respectively. Research and development expense in the three-month and six-month periods ended March 31, 2006 included share-based compensation of $186,000 and $262,000, respectively.
The fair value of each option award is estimated on the date of grant using a Black-Scholes-Merton option pricing model (“Black-Scholes model”) that uses assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, our expected stock price volatility, actual and projected employee stock option exercise behaviors, risk-free interest rate and expected dividends. Expected volatilities are based on historical volatility of our common stock and other factors. The expected term of options granted is based on analyses of historical employee termination rates and option exercises. The risk-free interest rates are based on the U.S. Treasury yield in effect at the time of the grant. Since we do not expect to pay dividends on our common stock in the foreseeable future, we estimated the dividend yield to be 0%. FAS 123R requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We estimate pre-vesting forfeitures based on our historical experience.
If factors change and we employ different assumptions in the application of FAS 123R in future periods, the compensation expense that we record under FAS 123R may differ significantly from what we have recorded in the current period. There is a high degree of subjectivity involved when using option pricing models to estimate share-based compensation under FAS 123R. Because changes in the subjective input assumptions can materially affect our estimates of fair values of our share-based compensation, in our opinion, existing valuation models, including the Black-Scholes and lattice binomial models, may not provide reliable measures of the fair values of our share-based compensation. Consequently, there is a risk that our estimates of the fair values of our share-based compensation awards on the grant dates may bear little resemblance to the actual values realized upon the exercise, early termination or forfeiture of those share-based payments in the future. Certain share-based payments, such as employee stock options, may expire worthless or otherwise result in zero intrinsic value as compared to the fair values originally estimated on the grant date and reported in our financial statements. Alternatively, values may be realized from these instruments that are significantly in excess of the fair values originally estimated on the grant date and reported in our financial statements. There is currently no market-based mechanism or other practical application to verify the reliability and accuracy of the estimates stemming from these valuation models, nor is there a means to compare and adjust the estimates to actual values. Although the fair value of employee share-based awards is determined in accordance with FAS 123R and the SEC’s Staff Accounting Bulletin No. 107 (“SAB 107”) using an option-pricing model, that value may not be indicative of the fair value observed in a willing buyer/willing seller market transaction.
28
The guidance in FAS 123 (R) and SAB 107 is relatively new, and best practices are not well established. The application of these principles may be subject to further interpretation and refinement over time. There are significant differences among valuation models, and there is a possibility that we will adopt different valuation models in the future. This may result in a lack of consistency in future periods and materially affect the fair value estimate of share-based payments. It may also result in a lack of comparability with other companies that use different models, methods and assumptions.
Theoretical valuation models and market-based methods are evolving and may result in lower or higher fair value estimates for share-based compensation. The timing, readiness, adoption, general acceptance, reliability and testing of these methods is uncertain. Sophisticated mathematical models may require voluminous historical information, modeling expertise, financial analyses, correlation analyses, integrated software and databases, consulting fees, customization and testing for adequacy of internal controls. Market-based methods are emerging that, if employed by us, may dilute our earnings per share and involve significant transaction fees and ongoing administrative expenses. The uncertainties and costs of these extensive valuation efforts may outweigh the benefits to investors.
For purpose of estimating the fair value of stock options granted during the six-month period ended March 31, 2006 using the Black-Scholes model, we have made an estimate regarding our stock price volatility (weighted average of 79.0%). If our stock price volatility assumption were increased to 89.0%, the weighted average estimated fair value per share of stock options granted during the six-month period ended March 31, 2006 would increase by $1.11, or 13%. The volatility percentage assumed in the six-month period ended March 31, 2006 was based on the historical prices of our common stock.
The expected term of options granted is based on our analyses of historical employee terminations and option exercises (weighted average of 4.5 years for the six-month period ended March 31, 2006) which, if increased to 5.5 years, would increase the weighted average estimated fair value per share of stock options granted during the six-month period ended March 31, 2006 by $1.10 or 13%.
The risk-free interest rate is based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of grant (weighted average of 4.4% for the six-month period ended March 31, 2006) which, if increased to 5.4%, would increase the weighted average estimated fair value per share of stock options granted during the six-month period ended March 31, 2006 by $0.50, or 6%.
The pre-vesting forfeiture rate is estimated using historical option cancellation information (weighted average of 8.0% for both officers and directors and 13% for employees for the six-month period ended March 31, 2006) which, if decreased to 3.0% for officers and directors and 8.0% for employees, would increase the share-based compensation expense for the six-month period ended March 31, 2006 by $96,000, or 8%. See Note 3, “Significant Accounting Policies — Change in Accounting Method for Share-Based Compensation,” in the Notes to Condensed Consolidated Financial Statements (Unaudited) for a detailed discussion.
Revenue Recognition
We allocate revenue to separate elements in multiple element arrangements based on the guidance in Emerging Issues Task Force No. 00-21 (“EITF 00-21”), “Accounting for Revenue Arrangements with Multiple Deliverables.” Revenues allocated to deliverables are recognized in accordance with SEC Staff Accounting Bulletin Topic 13 (“SAB Topic 13”), “Revenue Recognition” and revenue is recognized when four basic criteria of revenue recognition are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services rendered; (3) the fee is fixed or determinable; and (4) collectibility is reasonably assured.
Revenue Arrangements with Multiple Deliverables. We have revenue arrangements whereby we deliver to the customer multiple products and/or services (multiple deliverables). Such arrangements may include antibody generation services agreements, licensed compounds and intellectual property, and research and development services. In
29
accordance with EITF 00-21, we analyze our multiple element arrangements to determine whether the elements can be separated and accounted for individually as separate units of accounting. The evaluation is performed at the inception of the arrangement and as each element is delivered. The delivered item(s) is (are) considered a separate unit of accounting if all of the following criteria are met: (1) the delivered item(s) has (have) value to the customer on a standalone basis; (2) there is objective and reliable evidence of the fair value of the undelivered item(s); and (3) if the arrangement includes a general right of return relative to the delivered item(s), delivery, or performance of the undelivered item(s) is (are) considered probable and substantially in our control.
License Arrangements. License arrangements may consist of non-refundable upfront license fees, data transfer fees, or research reimbursement payments and/or exclusive licensed rights to patented or patent pending compounds, technology access fees, various performance or sales milestones and future product royalty payments. Such arrangements may also include fees for specific research and development services. If the delivered elements do not have standalone value or if we do not have objective or reliable evidence of the fair value of the undelivered component, the amount of revenue allocable to the delivered element is deferred. Non-refundable, up-front license fees, data transfer fees, and research reimbursement payments that have standalone value that are not contingent on any future performance by us, and that do not rely on our continuing involvement including any proprietary technology under the arrangements, and for which collection is assured, are recognized as revenue upon the delivery of the data and/or licensed compounds and commencement of the license term. Such deliverables may include physical quantities of compounds, design of the compounds and structure-activity relationships, the conceptual framework and mechanism of action, and rights to the patents or patents pending for such compounds. We consider licensed rights or licensed technology to have standalone value to our customers if we or others have sold such rights or technology separately or our customers can sell such rights or technology separately without the need for our continuing involvement. We defer revenue if we have continuing performance obligations without which the technology, right, product or service conveyed in conjunction with the non-refundable fee has no utility to the licensee that is separate and independent of our performance under the other elements of the arrangement. In addition, if we have continuing involvement through research and development services that are required because our know-how and expertise related to the technology is proprietary to us, or can only be performed by us, then such up-front fees are deferred and recognized over the period of continuing involvement.
Payments related to substantive, performance-based milestones are recognized as revenue upon the achievement of the milestones as specified in the underlying agreements, when they represent the culmination of the earnings process. Revenue from research and development services is recognized during the period in which the services are performed and is based upon the number of FTE personnel working on the specific project at the agreed-upon rate. Reimbursements from collaborative partners for agreed upon direct costs including direct materials and outsourced, or subcontracted, pre-clinical studies are recognized as revenues in the period the reimbursable expenses are incurred. Payments received in advance are recorded as deferred revenue until the research and development services are performed, costs are incurred, or a milestone is reached.
Royalty Revenues.We recognize royalty revenues from licensed products when earned in accordance with the terms of the license agreements. Net sales figures used for calculating royalties include deductions for costs of unsaleable returns, managed care chargebacks, cash discounts, freight and warehousing, and miscellaneous write-offs, which may vary over the course of the license agreement.
Revenues from Sale of Royalty Rights.In agreements where we have sold our rights to future royalties under license agreements and we maintain continuing involvement in earning such royalties, we defer revenues and recognize them over the life of the license agreement. For example, in the sale of an undivided interest of our Abreva® license agreement to Drug Royalty USA, revenue recognition is being determined under the units-of-revenue method. Under this method, the amount of deferred revenue to be recognized as revenue in each period is calculated by multiplying the following: (1) the ratio of the royalty payments due to Drug Royalty USA for the period to the total remaining royalties that we expect GlaxoSmithKline will pay Drug Royalty USA over the term of the agreement by (2) the unamortized deferred revenue amount.
30
Government Research Grant Revenue.We recognize revenues from federal research grants during the period in which the related expenditures are incurred.
Product Sales — Active Pharmaceutical Ingredient Docosanol.Revenue from product sales, which consist of sales of our active pharmaceutical ingredient docosanol, is recorded when the above criteria are met, generally when title and risk of loss have passed to the buyer upon delivery. We sell the active pharmaceutical ingredient docosanol to various licensees and ship only on written order for the materials. Total shipments generally occur fewer than five times a year. Our contracts for sales of the active pharmaceutical ingredient docosanol include buyer acceptance provisions that give our buyers the right of replacement if the delivered materials do not meet specified criteria by giving notice within 30 days after receipt of such defective materials. We have the option to refund or replace any such defective materials; however, we have historically demonstrated that the materials shipped from the same pre-inspected lot have consistently met the specified criteria and no buyer has rejected any of our shipments from the same pre-inspected lot to date. Therefore, we recognize revenue from sales of the active pharmaceutical ingredient docosanol without providing for such contingency upon shipment.
Recognition of Expenses in Outsourced Contracts
Pursuant to management’s assessment of the services that have been performed on clinical trials and other contracts, we recognize expenses as the services are provided. Such management assessments include, but are not limited to: (1) an evaluation by the project manager of the work that has been completed during the period, (2) measurement of progress prepared internally and/or provided by the third-party service provider, (3) analyses of data that justify the progress, and (4) management’s judgment. Several of our contracts extend across multiple reporting periods, including our largest contract, representing an $8.9 million Phase III clinical trial contract as of March 31, 2006. A 3% variance in our estimate of the work completed in our largest contract could increase or decrease our quarterly operating expenses by approximately $267,000.
Research and Development Expenses
Research and development expenses consist of expenses incurred in performing research and development activities including salaries and benefits, facilities and other overhead expenses, clinical trials, contract services and other outside expenses. Research and development expenses are charged to operations as they are incurred. Up-front payments to collaborators made in exchange for the avoidance of potential future milestone and royalty payments on licensed technology are also charged to research and development expense when the drug is still in the development stage, has not been approved by the FDA for commercialization and concurrently has no alternative uses.
We assess our obligations to make milestone payments that may become due under licensed or acquired technology to determine whether the payments should be expensed or capitalized. We charge milestone payments to research and development expense when:
| | • | | The technology is in the early stage of development and has no alternative uses; |
| |
| | • | | There is substantial uncertainty of the technology or product being successful; |
| |
| | • | | There will be difficulty in completing the remaining development; and |
| |
| | • | | There is substantial cost to complete the work. |
Acquired contractual rights. Payments to acquire contractual rights to a licensed technology or drug candidate are expensed as incurred when there is uncertainty in receiving future economic benefits from the acquired contractual rights. We consider the future economic benefits from the acquired contractual rights to a drug candidate to be uncertain until such drug candidate is approved by the FDA or when other significant risk factors are abated.
31
Capitalization and Valuation of Long-Lived and Intangible Assets
Intangible assets with finite useful lives consist of capitalized legal costs incurred in connection with patents, patent applications pending and license agreements. We amortize costs of approved patents, patent applications pending and license agreements over their estimated useful lives, or terms of the agreements, whichever are shorter. For patents pending, we amortize the costs over the shorter of a period of twenty years from the date of filing the application or, if licensed, the term of the license agreement. We re-assess the useful lives of patents when they are issued, or whenever events or changes in circumstances indicate the useful lives may have changed. For patent and patent applications pending and trademarks that we abandon, we charge the remaining unamortized accumulated costs to expense.
Intangible assets and long-lived assets are evaluated for impairment whenever events or changes in circumstances indicate that their carrying value may not be recoverable. If the review indicates that intangible assets or long-lived assets are not recoverable (i.e. the carrying amount is less than the future projected undiscounted cash flows), their carrying amount would be reduced to fair value. Factors we consider important that could trigger an impairment review include the following:
| | • | | A significant underperformance relative to expected historical or projected future operating results; |
| |
| | • | | A significant change in the manner of our use of the acquired asset or the strategy for our overall business; and/or |
| |
| | • | | A significant negative industry or economic trend. |
When we determine that the carrying value of intangible assets or long-lived assets are not recoverable based upon the existence of one or more of the above indicators of impairment, we may be required to record impairment charges for these assets.
RESULTS OF OPERATIONS
COMPARISON OF THREE MONTHS ENDED MARCH 31, 2006 AND 2005
Revenues
Total revenues increased to $2.5 million for the three-month period ended March 31, 2006, compared to $645,000 for the three-month period ended March 31, 2005. Revenues in the second quarter of fiscal 2006 included $2.0 million in research and development service revenues generated from our collaborative agreements with AstraZeneca and Novartis executed in July 2005 and April 2005, respectively, $427,000 from the recognition of revenue related to the sale of an undivided interest in our Abreva® license agreement to Drug Royalty USA and $76,000 from a government research grant. Revenue in the second quarter of fiscal 2005 included $420,000 from the recognition of deferred revenue, $100,000 relating to the achievement of a milestone under a license agreement and $125,000 from government research grants.
Revenue-generating contracts that remained active as of March 31, 2006 include license agreements with AstraZeneca and Novartis, eight docosanol 10% cream license agreements and one Neurodex™ sublicense. Partnering, licensing and research collaborations have been, and will continue to be, an important part of our business development strategy. We intend to partner with pharmaceutical companies that can help fund our research in exchange for sharing in the rights to commercialize new drugs resulting from this research. We have licensed and continue to seek licensees in other countries for docosanol 10% cream and other potential products in our development pipeline. Research collaborations also represent an important way to achieve our development goals, while sharing in the risks and the opportunities that come from such development efforts.
Operating Expenses
Total operating expenses increased by $1.6 million, or 11%, to $16.4 million for the three-month period ended March 31, 2006, compared to $14.8 million for the same period in fiscal 2005. The increase in operating expenses was caused by a $5.1 million, or 152%, increase in selling, general and administrative expenses; offset by a $3.5 million, or 31%, decrease in research and development expenses. These and other costs and trends are more fully described below.
| | | | | | | | | | | | | | | | | |
| | | Three Months Ended | | | Six Months Ended | |
| | | March 31, | | | March 31, | |
| | | 2006 | | | 2005 | | | 2006 | | | 2005 | |
| Operating expenses: | | | | | | | | | | | | | | | | |
| Research and development | | | 48 | % | | | 77 | % | | | 57 | % | | | 72 | % |
| Selling, general and administrative | | | 52 | % | | | 23 | % | | | 43 | % | | | 28 | % |
| Costs of sales | | | — | % | | | — | % | | | — | % | | | — | % |
| | | | | | | | | | | | | |
| Total operating expenses | | | 100 | % | | | 100 | % | | | 100 | % | | | 100 | % |
| | | | | | | | | | | | | |
32
Research and development (“R&D”) expenses.R&D expenses for the three-month periods ended March 31, 2006 and 2005 were $7.9 million and $11.4 million, respectively. R&D expenses in the second quarter of fiscal 2006 were related to a Phase III clinical trial of Neurodex for the treatment of painful diabetic neuropathy, continuation of the open label safety study of Neurodex in the treatment for IEED/PBA, and a Phase I clinical trial of our leading compound for the selective cytokine inhibitor. R&D expenses also included pre-clinical research related to antibody development programs and the compounds that regulate MIF and reverse cholesterol transport enhancing compounds; the development of these two compounds is funded by our collaborative partners. The higher R&D expenses in the three-month period ended March 31, 2005 were attributable to a $7.2 million charge related to the acquisition of additional contractual rights to Neurodex™ in that period. The period-over-period decrease is offset in part by a $2.4 million increase in spending for a Phase III clinical trial of Neurodex for the treatment of painful diabetic neuropathy and a $628,000 increase in pre-clinical development of our reverse cholesterol transport enhancing compounds. Approximately 23% of our program spending for the three-month period ended March 31, 2006 was in connection with research services funded by our partners; we received no such revenues from partners in the same period a year ago.
The following table sets forth the status of, and costs attributable to, our proprietary research and clinical development programs.
Research and Development Projects and Expenses
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Three Months Ended | | | Six Months Ended | | | Inception | | | Estimated | |
| | | March 31, | | | March 31, | | | Through | | | Cost to | |
| | | | | | | | | | | | | | | | | | | March 31, | | | License or Complete | |
| | | 2006 (1) | | | 2005 (1)(4) | | | 2006 (1) | | | 2005 (1)(4) | | | 2006 (1)(2)(4) | | | Project (1)(3) | |
Company-funded Projects: | | | | | | | | | | | | | | | | | | | | | | | | |
| Development programs for Neurodex, selective cytokine inhibitor and other programs projected through fiscal 2007 (3) | | $ | 6,027,247 | | | $ | 10,249,442 | | | $ | 13,304,575 | | | $ | 13,774,474 | | | $ | 92,044,454 | | | $30M to $40M |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Partner-funded Projects: | | | | | | | | | | | | | | | | | | | | | | | | |
| Reverse cholesterol transport and MIF inhibitor research programs | | | 1,795,634 | | | | 1,049,067 | | | | 3,811,719 | | | | 2,429,819 | | | | 23,267,609 | | | Funded by Partners |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Government-funded Projects: | | | | | | | | | | | | | | | | | | | | | | | | |
| Preclinical research on various projects. Funding for the anthrax research project has been | | | | | | | | | | | | | | | | | | | | | | | | |
| completed | | | 96,966 | | | | 129,661 | | | | 166,955 | | | | 278,118 | | | | 2,688,949 | | | $ | — | |
| | | | | | | | | | | | | | | | | | | | |
| | | $ | 7,919,847 | | | $ | 11,428,170 | | | $ | 17,283,249 | | | $ | 16,482,411 | | | $ | 118,001,012 | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | |
| (1) | | Each project includes an allocation of laboratory occupancy costs. “M” refers to millions. Estimated costs and timing to complete the projects are subject to the availability of funds. For each of the projects set forth in the table, other than Neurodex for IEED/PBA (which we intend to market ourselves), the reverse cholesterol transport and the MIF inhibitor programs (both of which we have partnered), we may seek development partners or licensees to defray part or all of the ongoing development costs. |
| |
| (2) | | Inception dates are on or after October 1, 1998, at which time we began identifying and tracking program costs. |
| |
| (3) | | Assumes completion of development of Neurodex in the treatment of IEED/PBA and for one Phase III clinical trial in the treatment of painful diabetic neuropathy and continuation of Phase I studies of our selective cytokine inhibitor program. Projected spending thereafter will be subject to progress made in research that is currently underway. |
| |
| (4) | | Includes expenses funded by us prior to partnering these projects in the amounts of $1.0 million and $2.4 million for the three-month and six-month periods ended March 31, 2005, respectively, and $17.1 million since inception through March 31, 2006. |
33
We expect that increases in R&D spending for painful diabetic neuropathy and potentially other indications will partially offset the expected decline in spending related to IEED/PBA in the coming years as we continue Phase III clinical trials of Neurodex in the treatment of painful diabetic neuropathy. All future R&D spending on compounds that regulate MIF and reverse cholesterol transport enhancing compounds is expected to be fully reimbursed by our collaborative partners. We expect that spending on our selective cytokine inhibitor program and on development of monoclonal antibodies will depend in part on the progress that we make in these programs and on our strategy for partnering these programs or in obtaining additional government grants, so that we are able to defray part or all of these ongoing development costs.
Status of R&D Programs and Plans — Company-funded Projects
Neurodex™ for the treatment of Involuntary Emotional Expression Disorder/Pseudobulbar Affect.We completed the submission of the Neurodex NDA to the FDA in January 2006, and the submission was accepted by the FDA for filing. As Neurodex received priority review status, we expect the FDA will take action on the NDA by July 30, 2006. However, this could be delayed if there are questions by the FDA about our NDA. We have been engaged in an open-label safety study for the treatment of IEED/PBA in a broad pool of patients who have IEED/PBA associated with their underlying neurodegenerative disease or condition. We expect to continue this open-label study until the FDA takes action on the NDA, plus any additional time until launch, if approved. In June 2004, we successfully completed the treatment phase of a Phase III clinical trial of Neurodex for the treatment of IEED/PBA in 150 patients with MS. Prior to engaging in these recent and current ongoing studies, we successfully completed the initial Phase III clinical trial of IEED/PBA in patients with ALS in May 2002.
Neurodex for the treatment of painful diabetic neuropathy.In June 2005, we initiated our first Phase III clinical trial of Neurodex in patients who have painful diabetic neuropathy and we are currently evaluating the balance of our development plan. Simultaneously, we are evaluating commercial development alternatives for this indication, including continuing development on our own (including conducting a second Phase III clinical trial) or co-promotion/licensing opportunities in which a partner would fund the second trial. Estimated timing to complete the first Phase III clinical trial is up to two years. There can be no assurances that we will choose or be able to negotiate a licensing and/or co-promotion arrangement for Neurodex in the treatment of painful diabetic neuropathy on attractive terms, if at all.
Development program for selective cytokine inhibitor.In November 2005, we completed a multi-rising dose Phase Ib safety trial of AVP-13358 and anticipate we will submit the results to the FDA in June 2006 for evaluation. We are currently evaluating several therapeutic indications for this program. In 2004 we completed the first Phase Ia clinical trial of AVP-13358 in 54 healthy volunteers. The placebo-controlled study was intended to assess safety, tolerability and pharmacokinetics following single rising oral doses. Results of the Phase Ia study suggest AVP-13358 was well tolerated at all single rising doses up through 15 milligrams. The study also demonstrated AVP-13358 was detectable in the bloodstream at all doses administered and remains in circulation long enough to allow potentially once or twice daily dosing.
34
Status of R&D Programs and Plans — Partner-funded Projects
Development program for reverse cholesterol transport.In July 2005 we entered into an exclusive license and research collaboration agreement with AstraZeneca to discover, develop and commercialize reverse cholesterol transport enhancing compounds for the treatment of cardiovascular disease. Under the terms of the agreement, we are eligible to receive royalty payments, assuming the product is successfully developed and approved for marketing by the FDA. We are also eligible to receive up to $330 million in milestone payments contingent upon achievement of certain development and regulatory milestones, which could take several years of further development, including achievement of certain sales targets, a licensed compound is approved for marketing by the FDA. Under the terms of the agreement, we received a license fee of $10.0 million in July 2005 and will also receive research funding of between $2.5 million and $4.0 million per year for providing research and development services to AstraZeneca for up to three years. AstraZeneca has the responsibility for all development costs. In December 2005, we submitted an IND to the FDA for the compound AZD 2479 (also known as AVP-26452) and began enrolling patients in a Phase I study to evaluate the safety of this compound, which triggered a $5.0 million milestone payment from AstraZeneca under the terms of the collaboration agreement. We expect spending on this program will remain about the same in the remaining quarters of fiscal 2006; however, AstraZeneca will be paying us for such services.
Anti-inflammatory research program (MIF inhibitor).In April 2005, we entered into a research, development and commercialization agreement with Novartis International Pharmaceutical Ltd. for orally active small molecule therapeutics that regulate MIF in the treatment of various inflammatory diseases. Under the terms of the agreement, we will be eligible to receive royalty payments, assuming the product is successfully developed and approved for marketing by the FDA. We are also eligible to receive up to $198 million in milestone payments contingent upon achievement of certain development, including approvals for certain additional indications, and regulatory milestones, which could take several years of further development, including achievement of certain sales targets, if a MIF compound is approved for marketing by the FDA. Under this agreement, we received a license fee of $2.5 million in May 2005 and will also receive research funding of between $1.5 million and $2.5 million per year for providing research and development services to Novartis for two years from the date of the agreement, or longer upon mutual agreement of the parties. Novartis has assumed responsibility for all development expenses.
Status of R&D Programs and Plans — Government-Funded Projects
Government research grants have helped us fund research programs, including the development of antibodies to anthrax toxins and docosanol-based formulations for the treatment of genital herpes. Subject to certain conditions, we, as the awardee organization, retain the principal worldwide patent rights to any invention developed with the United States government support.
Two of our most potent anthrax antibodies, AVP-21D9 and AVP-22G12, appear unique both in mechanism of action and the binding site on an anthrax toxin. AVP-21D9 was in preclinical development for use as a prophylactic and therapeutic drug to treat anthrax infections and was being funded by a two-year $750,000 federal (SBIR) research grant. The grant was completed in the quarter ended March 31, 2006. Much of the work related to anthrax had been funded by government research grants, and our progress in this area will substantially depend on future grants. Because all of our monoclonal antibody research is at a very early preclinical stage of development and is unpredictable in terms of the outcome, we are unable to predict the cost and timing for development of any antibody or drug.
35
Our genital herpes project came to a formal end during the third quarter of fiscal 2005 and we do not anticipate that we will perform any further work on that project. We have no grant requests pending nor do we anticipate submitting in the future any grant requests for further research related to genital herpes.
Selling, general and administrative expenses.Our selling, general and administrative expenses increased to $8.5 million for the three-month period ended March 31, 2006, compared to $3.4 million for the three-month period ended March 31, 2005. These increased expenses primarily relate to:
| | • | | $2.0 million increase in expenses related to the continued expansion of our market research and pre-launch planning activities for Neurodex for the treatment of IEED/PBA and hiring of additional sales and marketing personnel; |
| |
| | • | | $1.5 million in continuing medical education grants for IEED; |
| |
| | • | | $604,000 increase in expenses related to increases in headcount and compensation levels in general and administrative areas; and |
| |
| | • | | $572,000 share-based compensation expense. |
Based on our current commercial development plans for Neurodex for the treatment of IEED/PBA, we expect sales and marketing expenses in fiscal 2006 will continue to increase, although the timing and pace of these increases is difficult to predict and will be subject to fluctuation depending on the action taken by the FDA on our NDA later this year. We expect that costs related to professional services associated with compliance with the Sarbanes-Oxley Act of 2002 will decline from fiscal 2005 levels, although we expect offsetting increases in connection with compliance with healthcare laws and regulations.
Share-Based Compensation
Through fiscal 2005, we accounted for our stock plans using the intrinsic value method. Effective at the beginning of fiscal 2006, we adopted FAS 123R and elected to adopt the modified prospective application method. FAS 123R requires us to use a fair-valued based method to account for share-based compensation. Accordingly, share-based compensation cost is measured at the grant date, based on the fair value of the award, and is recognized as expense over the employees’ requisite service period. Total compensation cost for our share-based payments in the three-month period ended March 31, 2006 was $758,000. Selling, general and administrative expense and research and development expense in the three-month period ended March 31, 2006 include share-based compensation of $572,000 and $186,000, respectively.
Interest Income
For the three-month period ended March 31, 2006, interest income increased to $564,000, compared to $125,000 for the same period in the prior year. The increase is primarily due to a 141% increase in average balance of cash, cash equivalents and investments in securities for the second quarter of fiscal 2006, compared to the same period in the prior year.
Net Loss
Net loss was $13.4 million, or $0.43 per share, in the three-month period ended March 31, 2006, compared to a net loss of $14.1 million, or $0.57 per share, in the three-month period ended March 31, 2005. A lower net loss and a higher weighted average number of shares outstanding in the second quarter of fiscal 2006 accounted for the lower loss per share.
We expect to continue to pursue our drug development strategy focused on the development of Neurodex™, followed by other programs in earlier stages of development that are in large therapeutic areas and that have significant partnering and licensing potential. Effective April 2005, Novartis assumed all responsibility for ongoing product development expenses for our compounds that regulate MIF in the
36
treatment of various inflammatory diseases. Effective July 2005, AstraZeneca assumed all responsibility for ongoing product development expenses for our reverse cholesterol transport enhancing compounds to reduce cardio vascular disease. To help fund and develop our other product candidates, we may elect to license those technologies and drug candidates to help offset the costs of development. These potential license arrangements could materially change our outlook for future revenues and costs. However, the timing of such potential arrangements is unpredictable.
COMPARISON OF SIX-MONTH PERIODS ENDED MARCH 31, 2006 AND 2005
Revenues
Total revenues increased to $10.6 million for the six-month period ended March 31, 2006, compared to $1.5 million for the six-month period ended March 31, 2005. Revenues in the first half of fiscal 2006 included $5.0 million relating to the achievement of a milestone under the AstraZeneca license agreement, $4.4 million in research and development service revenues generated from our collaborative agreements with AstraZeneca and Novartis executed in July 2005 and April 2005, respectively, and $1.0 million from the recognition of revenue related to the sale of an undivided interest in our Abreva® license agreement to Drug Royalty USA. Revenue in the first half of fiscal 2005 included $930,000 from the recognition of deferred revenue, and $300,000 relating to the achievement of milestones under license agreements.
Operating Expenses
Total operating expenses increased by $7.7 million, or 34%, to $30.5 million for the six-month period ended March 31, 2006, compared to $22.8 million for the same period in fiscal 2005. The increase in operating expenses was caused by a $801,000, or 5%, increase in research and development expenses and a $6.9 million, or 110%, increase in selling, general and administrative expenses.
Research and development (“R&D”) expenses.R&D expenses for the six-month periods ended March 31, 2006 and 2005 were $17.3 million and $16.5 million, respectively. R&D expenses in the first half of fiscal 2006 were related to continuation of the open label safety study of Neurodex™ in the treatment for IEED/PBA, a Phase III clinical trial of Neurodex for the treatment of painful diabetic neuropathy, and a Phase I clinical trial of our leading compound for the selective cytokine inhibitor program. R&D expenses also included pre-clinical research related to antibody development programs and compounds that regulate MIF and reverse cholesterol transport enhancing compounds; the development of these two compounds is funded by our partners. The increase in R&D expenses is due to a $1.9 million increase in spending for the open label safety study of Neurodex for the treatment of IEED/PBA, a $3.9 million increase in spending for a Phase III clinical trial of Neurodex for the treatment of painful diabetic neuropathy and a $1.1 million increase in pre-clinical development of our reverse cholesterol transport. Approximately 22% of our program spending for the six-month period ended March 31, 2006 was in connection with research services funded by our partners; we received no such revenues from partners in the same period in the prior year. The period-over-period increase was offset by the one-time $7.2 million charge incurred in connection with the acquisition of additional contractual rights to Neurodex in the prior year.
Selling, general and administrative expenses.Our selling, general and administrative expenses increased to $13.3 million for the six-month period ended March 31, 2006, compared to $6.3 million for the six-month period ended March 31, 2005. These increased expenses primarily relate to:
| | • | | $2.8 million increase in expenses related to the continued expansion of our market research and pre-launch planning activities for Neurodex for the treatment of IEED/PBA and hiring of additional sales and marketing personnel; |
| |
| | • | | $1.5 million in continuing medical educational grants; |
37
| | • | | $1.1 million increase in expenses related to increases in headcount and compensation levels in general and administrative areas; and |
| |
| | • | | $957,000 share-based compensation expense. |
Share-Based Compensation
Total compensation cost for our share-based payments in the six-month period ended March 31, 2006 was $1.2 million. Selling, general and administrative expense and research and development expense in the six-month period ended March 31, 2006 include share-based compensation of $957,000 and $262,000, respectively.
Interest Income
For the six-month period ended March 31, 2006, interest income increased to $893,000, compared to $247,000 for the same period in the prior year. The increase is primarily due to a 90% increase in average balance of cash, cash equivalents and investments in securities for the first half of fiscal 2006, compared to the same period in the prior year.
Net Loss
Net loss was $19.1 million, or $0.64 per share, in the six-month period ended March 31, 2006, compared to a net loss of $21.1 million, or $0.87 per share, in the six-month period ended March 31, 2005. A lower net loss and a higher weighted average number of shares outstanding in the first half of fiscal 2006 accounted for the lower loss per share.
LIQUIDITY AND CAPITAL RESOURCES
As of March 31, 2006, we had cash, cash equivalents, investments in securities and restricted investments totaling $53.0 million, including cash and cash equivalents of $10.9 million, short- and long-term investments of $41.2 million and restricted investments in securities of approximately $857,000. Our net working capital balance as of March 31, 2006 was $38.6 million. As of September 30, 2005, we had cash, cash equivalents, investments in securities and restricted investments totaling $27.5 million, including cash and cash equivalents of $8.6 million, short- and long-term investments of $18.1 million, and restricted investments of approximately $857,000. Our net working capital balance as of September 30, 2005 was $12.0 million. Explanations of net cash provided by or used for operating, investing and financing activities are provided in the table below.
| | | | | | | | | | | | | |
| | | | | | | Increase (Decrease) | | | September 30, | |
| | | March 31, 2006 | | | During Period | | | 2005 | |
| Cash, cash equivalents, investment in securities and restricted investments | | $ | 52,954,256 | | | $ | 25,416,670 | | | $ | 27,537,586 | |
| Cash and cash equivalents | | $ | 10,912,580 | | | $ | 2,292,437 | | | $ | 8,620,143 | |
| Net working capital | | $ | 38,602,472 | | | $ | 26,633,022 | | | $ | 11,969,450 | |
| | | | | | | | | | | | | |
| | | | | | | Change | | | | |
| | | Six Months Ended | | | Between | | | Six Months Ended | |
| | | March 31, 2006 | | | Periods | | | March 31, 2005 | |
| Net cash used for operating activities | | $ | (17,326,116 | ) | | $ | (2,065,340 | ) | | $ | (15,260,776 | ) |
| Net cash (used for) provided by investing activities | | | (24,595,598 | ) | | | (27,466,911 | ) | | | 2,871,313 | |
| Net cash provided by financing activities | | | 44,214,151 | | | | 37,112,285 | | | | 7,101,866 | |
| | | | | | | | | | |
| Net increase (decrease) in cash and cash equivalents | | $ | 2,292,437 | | | $ | 7,580,034 | | | $ | (5,287,597 | ) |
| | | | | | | | | | |
Operating activities.Net cash used for operating activities amounted to $17.3 million in the first half of fiscal 2006, $2.1 million higher than the net cash used for operating activities in the first half of fiscal 2005. The increase in cash used for operating activities is due to spending on an open label study and
38
consulting fees relating to Neurodex™ in the treatment of IEED/PBA and a Phase III clinical study for Neurodex in the treatment of painful diabetic neuropathy. The increase in cash used is also due to the continued expansion of our medical education and awareness programs for IEED/PBA, market research, and pre-launch activities for Neurodex, in anticipation of the drug being approved by the FDA. Based on our current commercial development plans for Neurodex for the treatment of IEED/PBA, we expect sales and marketing expenses will continue to increase, although the timing and rate of increase will depend on the response of the FDA to our NDA for Neurodex. We expect R&D spending will continue at about the same rate or slightly lower for the remainder of fiscal 2006, including clinical development of Neurodex in the treatment of painful diabetic neuropathy and the Phase III clinical trial. Additionally, we expect to continue spending at about the same rate on preclinical and clinical research services related to the development of compounds for the MIF and RCT programs in fiscal 2006. AstraZeneca and Novartis are funding the RCT and MIF programs, respectively, and pay us for these research services.
Accounts payable decreased by $3.5 million to $3.3 million at March 31, 2006 from $6.8 million at September 30, 2005, primarily due to payments of invoices in connection with our medical education and awareness programs and market research regarding IEED/PBA. A $3.1 million increase in accrued expenses to $7.2 million at March 31, 2006 from $4.1 million at September 30, 2005 resulted from accruals for goods and services received in the six-month period ended March 31, 2006 but were not yet invoiced by the vendors.
Investing activities.Net cash used for investing activities was $24.6 million in the first half of fiscal 2006, compared to $2.9 million provided by investing activities in the first quarter of fiscal 2005. Our investments in securities increased by $23.3 million in the first half of fiscal 2006 and decreased by $3.6 million in the first half of fiscal 2005, net of proceeds from sales and maturities of investments in securities. We invested $951,000 in property and equipment in the first half of fiscal 2006, compared to $151,000 in the first half of fiscal 2005. The increased spending was related primarily to product tooling, additional computer equipment, and in making improvements in office space utilization to accommodate additional personnel within existing leased space. We expect that capital expenditures for property and equipment will likely increase as we make further modifications to improve office space utilization and to make accommodations for additional sales and marketing personnel that will be necessary to support commercialization of Neurodex™, assuming the drug is approved by the FDA. (See Management’s Discussion and Analysis of Financial Condition and Results of Operations, “Selling, General and Administrative Expenses.”)
Financing activities.Net cash provided by financing activities was $44.2 million in the first half of fiscal 2006, consisting of $35.6 million in net proceeds from sales of our common stock through private placements and $8.7 million from exercises of warrants and stock options to purchase our common stock. Net cash provided by financing activities amounted to $7.1 million in the first half of fiscal 2005, consisting of $7.0 million received from the sale of our common stock and $100,000 from exercises of stock options.
In June 2005, we filed shelf registration statement on Form S-3 with the SEC to sell an aggregate of up to $100 million in Class A common stock and preferred stock, depositary shares, debt securities and warrants. This shelf registration statement was declared effective on August 3, 2005. In October 2005, we sold 1,523,585 shares of Class A common stock to certain institutional investors at $10.60 per share for aggregate net offering proceeds of $16.15 million. In December 2005, we sold 1,492,538 shares of our Class A common stock to certain institutional investors at $13.40 per share, for aggregate offering proceeds of approximately $20.0 million and net offering proceeds of approximately $19.4 million, after deducting commissions and offering fees and expenses.
Between January 26, 2006 and February 7, 2006, we received proceeds of $4.7 million from the exercise of warrants to purchase 671,923 shares of Class A common stock in connection with our call for redemption of a group of outstanding warrants. The warrants had been issued in connection with a financing transaction in December 2003 involving the sale of Class A common stock and warrants (the
39
“Warrants”). The exercise price of the Warrants was $7.00 per share. The Warrants had a five-year term, but included a provision that we could redeem the Warrants for $1.00 each if our stock price traded above twice the warrant exercise price for a certain period of time (the “Redemption Right”). On January 24, 2006, we sent the Warrant holders notice that the Redemption Right had been triggered and that the Warrants would expire, to the extent unexercised, on February 7, 2006. One of the warrants to purchase 25,167 shares of Class A common stock expired unexercised.
In September 2004, we entered into an equipment line of credit with GE Healthcare Financial Services for financing of up to $1.4 million. As of March 31, 2006, the outstanding balance due under the line of credit was approximately $800,000.
We hold the exclusive worldwide marketing rights to Neurodex™ for certain indications pursuant to an exclusive license agreement with the Center for Neurologic Study (“CNS”). We are currently developing Neurodex for the treatments of IEED and painful diabetic neuropathy. We will be obligated to pay CNS up to $400,000 in the aggregate in milestones to continue to develop both indications, assuming they are both approved for marketing by the FDA. We are not currently developing, nor do we have an obligation to develop, any other indications under the CNS license agreement. In fiscal 2005, we paid $75,000 to CNS under the CNS license agreement, and we expect to pay a $75,000 milestone in 2006 if the FDA approves our NDA for Neurodex for the treatment of IEED. In addition, we are obligated to pay CNS a royalty on commercial sales of Neurodex with respect to each indication, if and when the drug is approved by the FDA for commercialization. Under certain circumstances, we may have the obligation to pay CNS a portion of net revenues received if we sublicense Neurodex to a third party. We do not expect to pay any other milestones to CNS in fiscal 2006.
MANAGEMENT OUTLOOK
We believe that cash, cash equivalents, investments in securities and restricted investments of approximately $53.0 million at March 31, 2006, plus anticipated future cash from licensed technologies, should be sufficient to sustain our planned level of operations for at least the next 12 months. During fiscal 2006, we expect to earn $6.0 million to $8.0 million for the year in revenues from R&D services that we are providing under collaborative agreements. These payments will fully offset the expenses that we incur in connection with providing those services. During the remainder of fiscal 2006, we do not expect to earn any additional milestones under the AstraZeneca or the Novartis license agreement. In general, potential milestone payments to be received under existing license agreements are outside of our control and are far less predictable. Fiscal 2006 revenues from new sources, such as license fees and milestone payments, will depend substantially on whether or not we enter into additional license arrangements and whether or not we achieve milestones under those arrangements. Such arrangements could be in the form of licensing or partnering agreements for docosanol 10% cream, Neurodex, or for our other product development programs including development of a selective cytokine inhibitor. Many of our product development programs could take years of additional development before they reach the stage of being licensable to other pharmaceutical companies.
We expect to raise additional capital to support the launch and marketing of Neurodex within several months of the drug being reviewed by FDA for marketing. Potential alternatives that we could pursue for raising capital include, but are not limited to, partnering arrangements where partners share development costs, issuance of debt or equity securities, and licensing or sales of one or more of our platform technologies or new drug candidates. The balance of securities available for sale under the existing shelf registration is approximately $64.0 million as of March 31, 2006. If we are unable to raise capital as needed to fund our operations, or if we are unable to reach milestones in our license agreements or enter into any new collaborative arrangements, then we may need to slow the rate of development of some of our programs or sell the rights to one or more of our drug candidates. For information regarding the risks associated with our need to raise capital to fund our ongoing and planned operations, please see “Risk Factors.”
40
RECENT ACCOUNTING PRONOUNCEMENTS
See Note 3, “Significant Accounting Policies — Change in Accounting Method for Share-based Compensation” and Note 18, “Recent Accounting Pronouncements” in the Notes to Condensed Consolidated Financial Statements (Unaudited) for a discussion of recent accounting pronouncements and their effect, if any, on our financial condition or results of operations.
Item 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
As described below, we are exposed to market risks related to changes in interest rates. Because substantially all of our revenue, expenses, and capital purchasing activities are transacted in U.S. dollars, our exposure to foreign currency exchange rates is immaterial. However, in the future we could face increasing exposure to foreign currency exchange rates as we expand international distribution of docosanol 10% cream and purchase additional services from outside the U.S. Until such time as we are faced with material amounts of foreign currency exchange rate risks, we do not plan to use derivative financial instruments, which can be used to hedge such risks. We will evaluate the use of derivative financial instruments to hedge our exposure as the needs and risks should arise.
Interest rate sensitivity
Our investment portfolio consists primarily of fixed income instruments with an average duration of 0.7 year as of March 31, 2006 (1.2 years as of September 30, 2005). The primary objective of our investments in debt securities is to preserve principal while achieving attractive yields, without significantly increasing risk. We classify our investments in securities as of March 31, 2006 as available-for-sale and our restricted investments in securities as held-to-maturity. These available-for-sale securities are subject to interest rate risk. In general, we would expect that the volatility of this portfolio would decrease as its duration decreases. Based on the average duration of our investments as of March 31, 2006 and 2005, an increase of one percentage point in the interest rates would have resulted in increases in comprehensive losses of approximately $372,000 and $162,000, respectively.
Item 4. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our chief executive officer and chief financial officer, after evaluating the effectiveness of our disclosure controls and procedures (as defined in the Securities Exchange Act of 1934, Rules 13a-15(e) and 15d-15(e), as of the end of the period covered by this report, have concluded that, based on such evaluation, as of March 31, 2006 our disclosure controls and procedures were effective and designed to provide reasonable assurance that the information required to be disclosed is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission rules and forms. In designing and evaluating the disclosure controls and procedures, our management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Changes in Internal Controls over Financial Reporting
There has been no change in the Company’s internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) during the Company’s fiscal quarter ended March 31, 2006, that has materially affected, or is reasonably likely to materially affect the Company’s internal control over financial reporting.
41
PART II OTHER INFORMATION
Item 1. LEGAL PROCEEDINGS
In the ordinary course of business, we may face various claims brought by third parties. Any of these claims could subject us to costly litigation. Management believes the outcome of currently identified claims and lawsuits will not have a material adverse effect on our financial condition or results of operations.
Item 1A. RISK FACTORS
Risks Relating to Our Business
We have a history of losses and we may never achieve or maintain profitability.
We have experienced significant operating losses in funding the research, development and clinical testing of our drug candidates, accumulating operating losses totaling $174.1 million as of March 31, 2006, and we expect to continue to incur substantial operating losses through at least fiscal 2007. To achieve profitability, we will need to generate significant additional revenue with positive gross margins to offset our other operating expenses, which we expect will increase as we pursue pre-launch activities and sales and marketing efforts over the next several quarters in anticipation of a favorable the FDA decision regarding the approval of Neurodex™ for the treatment of IEED/PBA.
We may also increase spending on our clinical and pre-clinical programs to the extent our progress in development is favorable. Although we continue to seek new revenue-generating licenses for docosanol 10% cream and development partnerships for certain of our other research programs, including for a selective cytokine inhibitor and our antibody technology, we may not find additional attractive arrangements, if at all, and any such arrangements may not provide adequate revenues to cover future operating expenses. Increases in expenditures may not be offset by new or adequate sources of revenues, and as a result, we may not achieve profitability.
Any adverse decisions in the regulatory review or approval process may harm our prospects and could harm our stock price.
In April 2006, we announced that the FDA accepted our NDA submission for Neurodex with priority review and we expect that the FDA will take action on the NDA by July 30, 2006. We cannot be certain that Neurodex will be approved by the FDA for marketing or that we will be able to obtain the labeling claims that we consider most desirable for the promotion of the product. Additionally, recent announcements by others regarding safety problems with certain approved drugs may affect the FDA’s policies regarding safety data for all new drug applications and may result in the FDA requiring additional safety data before approving Neurodex and/or the FDA requiring closer surveillance after commercialization if the drug is approved.
We have yet to market or sell Neurodex or any of our other potential products.
We have never before directly marketed or sold any pharmaceutical products. In order to market Neurodex, assuming it is approved by the FDA, or market any other drug candidates, we will need to hire additional personnel with relevant pharmaceutical experience to staff our sales management and marketing group. If we cannot develop the required marketing and sales expertise internally, our ability to generate revenue from product sales will likely suffer.
In international markets, we may rely on collaborative partners to obtain regulatory approvals and to market and sell our products in those markets. We have not yet entered into any collaborative arrangement with respect to marketing or selling Neurodex, with the exception of one such agreement relating to Israel. We cannot guarantee that we will be able to enter into any other arrangements on terms favorable to us, or at all. If we are able to enter into sales and marketing arrangements with collaborative partners, we cannot assure you that such collaborators will apply adequate resources and skills to their responsibilities, or that their marketing efforts will be successful. If we are unable to enter into favorable collaborative arrangements with respect to marketing or selling Neurodex or docosanol 10% cream, or if our collaborators’ efforts are unsuccessful, our ability to generate revenue from product sales will suffer.
42
We expect that we will need to raise additional capital to fund ongoing operations and support our planned product launch. If we are unable to raise additional capital, we may be forced to curtail operations. If we succeed in raising additional capital through a licensing or financing transaction, it may affect our stock price and future revenues.
In order to maintain sufficient cash and investments to fund future operations and to prepare for the commercialization of Neurodex™, we anticipate that we will need to raise additional capital over the next 12 to 24 months and that we may do so through various financing alternatives. These alternatives include licensing or sales of our technologies and drug candidates, selling shares of our Class A common stock or preferred stock, or through the issuance of one or more forms of senior or subordinated debt. The balance of securities available for sale under our existing shelf registration was approximately $64.0 million as of March 31, 2006. See “Management Outlook” in Management’s Discussion and Analysis of Financial Condition and Results of Operations.
Raising capital by issuing additional shares of Class A common stock may depress the market price of our Class A common stock and any such financing will dilute voting rights of our other shareholders. If we raise capital through licensing or sales of one or more of our technologies or drug candidates, as we have with our RCT and MIF technologies, then we will likely need to share a significant portion of future revenues from these drug candidates with our licensees. Additionally, the development of any drug candidates licensed or sold to third parties will no longer be in our control and thus we may not realize the full value of any such relationships.
If we are unable to raise additional capital to fund future operations, then we may not be able to execute our commercialization plans for Neurodex and may be required to reduce operations or defer or abandon one or more of our clinical or pre-clinical research programs.
Changes in board and management composition that are intended to strengthen the board and management team could adversely disrupt our operations.
We have recently made significant changes to our senior management team and board of directors to add to our pharmaceutical experience, significantly enhance our scientific and clinical expertise, and provide depth in managing profitable pharmaceutical businesses. Our President and Chief Executive Officer, Senior Vice President of Sales and Marketing, Vice President of Medical Affairs and Vice President of Sales have all joined the Company in the past 12 months. We also announced the appointment of a new Chief Financial Officer in May 2006. Since September 2004, seven new directors have joined our board and we continue to recruit senior-level personnel to add to our management team. Accordingly, we anticipate that we could experience other changes in management and infrastructure as we expand our organization, prepare for the commercialization of Neurodex and effect our transition from a research and development company to a pharmaceutical company. These changes could be disruptive, and we may experience difficulties in retaining new directors, attracting and integrating new members of the management team and in transitioning our operating activities. Any significant disruptions as a result of these changes could negatively impact our planned commercial launch of Neurodex.
Our inability to attract and retain key management and scientific personnel could negatively affect our business.
The industry in which we compete has a high level of employee mobility and aggressive recruiting of skilled personnel. This type of environment creates intense competition for qualified personnel, particularly in product research and development, sales and marketing and accounting and finance. The loss of certain executive officers and other key employees could adversely affect our operations.
Additionally, in order to expand the Company as planned and to effect our transition to a pharmaceutical company, we will need to hire, train, retain and motivate high quality personnel, including sales and marketing personnel for the commercialization of Neurodex. Any inability to hire qualified sales and marketing personnel would harm our commercialization plans. In May 2006, we announced that we would be moving our commercial and general and administrative operations from San Diego to Orange County, California. This move, and having operations in two locations, could impact our ability to retain and attract personnel. If we were to lose one or more of our key scientists, then we would likely lose some portion of our institutional knowledge and technical know-how, which could potentially cause a substantial delay in one or more of our development programs until adequate replacement personnel could be hired and trained.
43
We generally do not control the development of compounds licensed to third parties and, as a result, we may not realize a significant portion of the potential value of any such license arrangements.
Under our license arrangements for our RCT and MIF compounds, we have no direct control over the development of these drug candidates and have only limited input on the direction of development efforts. Because much of the potential value of these license arrangements is contingent upon the successful development and commercialization of the licensed technology, the ultimate value of these licenses will depend largely on the efforts of our licensing partners. If our licensing partners do not succeed in developing the licensed technology for whatever reason, we may be unable to realize a significant portion of the potential value of these arrangements.
Our patents may be challenged and our pending patents may be denied. Either result would seriously jeopardize our ability to compete in the intended markets for our proposed products.
We have invested in an extensive patent portfolio and we rely substantially on the protection of our intellectual property through our ownership or control of issued patents and patent applications. Such patents and patents pending cover Neurodex™, docosanol 10% cream and other potential drug candidates that could come from our technologies such as reverse cholesterol transport, selective cytokine inhibitors, anti-inflammatory compounds and antibodies. Because of the competitive nature of the biopharmaceutical industry, we cannot assure you that:
| | • | | The claims in any pending patent applications will be allowed or that patents will be granted; |
| |
| | • | | Competitors will not develop similar or superior technologies independently, duplicate our technologies, or design around the patented aspects of our technologies; |
| |
| | • | | Our proposed technologies will not infringe other patents or rights owned by others, including licenses that may be not be available to us; |
| |
| | • | | Any of our issued patents will provide us with significant competitive advantages; or |
| |
| | • | | Challenges will not be instituted against the validity or enforceability of any patent that we own or, if instituted, that these challenges will not be successful. |
Even if we successfully preserve our intellectual property rights, other biotechnology or pharmaceutical companies may allege that our technology infringes on their rights. Intellectual property litigation is costly, and even if we were to prevail in such a dispute, the cost of litigation could adversely affect our business, financial condition, and results of operations. Litigation is also time-consuming and would divert management’s attention and resources away from our operations and other activities. If we were to lose any litigation, in addition to any damages we would have to pay, we could be required to stop the infringing activity or obtain a license. Any required license might not be available to us on acceptable terms, or at all. Some licenses might be non-exclusive, and our competitors could have access to the same technology licensed to us. If we were to fail to obtain a required license or were unable to design around a competitor’s patent, we would be unable to sell or continue to develop some of our products, which would have a material adverse effect on our business, financial condition and results of operations.
We depend on third parties to manufacture, package and distribute compounds for our drugs and drug candidates. The failure of these third parties to perform successfully could harm our business.
We have utilized, and intend to continue utilizing, third parties to manufacture, package and distribute docosanol 10% cream, Neurodex and active pharmaceutical ingredients and supplies for our other drug candidates. We have no experience in manufacturing and do not have any manufacturing facilities. Currently, we have sole suppliers for the active pharmaceutical ingredients (“API”) for docosanol and Neurodex™, and a sole manufacturer for the finished form of Neurodex. Further, we do not have any long-term agreements in place with our current docosanol supplier or Neurodex API suppliers. Any delays or difficulties in obtaining API or in manufacturing, packaging or distributing Neurodex could delay our clinical trials for painful diabetic neuropathy and delay the commercialization of Neurodex for IEED/PBA. Additionally, the third parties we rely on for manufacturing and packaging are subject to regulatory review; and any regulatory compliance problems with these third parties could significantly delay or disrupt our commercialization activities.
44
Because we depend on clinical research centers and other contractors for clinical testing and for certain research and development activities, the results of our clinical trials and such research activities are, to a certain extent, beyond our control.
The nature of clinical trials and our business strategy of outsourcing a substantial portion of our research require that we rely on clinical research centers and other contractors to assist us with research and development, clinical testing activities, patient enrollment and regulatory submissions to the FDA. As a result, our success depends partially on the success of these third parties in performing their responsibilities. Although we pre-qualify our contractors and we believe that they are fully capable of performing their contractual obligations, we cannot directly control the adequacy and timeliness of the resources and expertise that they apply to these activities. If our contractors do not perform their activities in an adequate or timely manner, the pace of clinical development, regulatory approval and commercialization of our drug candidates could have to be abandoned or delayed.
Accounting for material transactions can be very complex and we face risks if regulators disagree with our accounting treatment for any of these transactions.
Many of the transactions that we engage in are recorded in our financial statements using complex accounting rules. As we prepare for the planned commercial launch of Neurodex, we expect that our financial accounting, including revenue recognition, will become even more complex. Although we believe that we have historically accounted for material transactions in accordance with generally accepted accounting principles (“GAAP”), our financial statements and related disclosure are subject to periodic reviews by the Staff of the Securities and Exchange Commission (“SEC”). The SEC is responsible for the final interpretation of GAAP and may ultimately disagree with our historic accounting practices.
During the three months ended March 31, 2006, the SEC provided comments on our Annual Report on Form 10-K for the year ended September 30, 2005 as part of the SEC’s regular review of the Company’s periodic reports and financial statements. We are currently in the process of researching and responding to these comments, which principally relate to the Company’s accounting and disclosure for certain transactions that occurred during fiscal 2005. The SEC’s comments relate only to non-cash accounting entries and do not affect our cash flows. Although we believe that we have properly accounted for these transactions, the SEC may ultimately disagree with our accounting treatment, in which case we may be required to restate certain financial statements in prior periods in accordance with the SEC’s requirements. Any such restatement could have a negative impact on our stock price and negatively affect the credibility of our financial reporting in future periods. Additionally, a restatement could negatively affect the assessment of the effectiveness of our internal controls over financial reporting.
Developing new pharmaceutical products for human use involves product liability risks, for which we currently have limited insurance coverage.
The testing, marketing and sale of pharmaceutical products involves the risk of product liability claims by consumers and other third parties. We maintain product liability insurance coverage for our clinical trials in the amount of $5 million per incident and $5 million in the aggregate. We expect to increase this coverage if we receive marketing approval for Neurodex by the FDA. However, product liability claims can be high in the pharmaceutical industry and our insurance may not sufficiently cover our actual liabilities. Additionally, our insurance carriers may deny, or attempt to deny, coverage in certain instances. If a suit against our business or proposed products is successful, then the lack of or insufficiency of insurance coverage could affect materially and adversely our business and financial condition. Furthermore, various distributors of pharmaceutical products require minimum product liability insurance coverage before their purchase or acceptance of products for distribution. Failure to satisfy these insurance requirements could impede our ability to achieve broad distribution of our proposed products.
45
Risks Relating to Our Stock
Our stock price has historically been volatile and we expect that this volatility may continue for the foreseeable future.
The market price of our Class A common stock has been, and is likely to continue to be, highly volatile. This volatility can be attributed to many factors, including the following:
| | • | | Comments made by securities analysts, including changes in their recommendations; |
| |
| | • | | Announcements by us of financing transactions and/or future sales of equity or debt securities; |
| |
| | • | | Sales of our Class A common stock by our directors, officers, or significant shareholders; |
| |
| | • | | Announcements by our competitors of clinical trial results or product approvals; |
| |
| | • | | Changes in operations and increases in operating expenses as we prepare for the commercial launch of Neurodex™, assuming it is approved by the FDA; and |
| |
| | • | | Market and economic conditions. |
Additionally, our stock price has been volatile as a result of periodic variations in our operating results. We expect our operating results to continue to vary from quarter-to-quarter due to the foregoing factors, as well as other factors, and these variations may be significant. Variations may result from the following factors:
| | • | | Performance under partnering arrangements— The recognition of the revenue under our partnering arrangements, including our license agreements for our MIF and RCT technologies, will partially depend on the efforts and performance of our licensees in reaching milestones that are outside of our control, such as regulatory approval, product launch, or reaching a sales threshold. |
| |
| | • | | Timing of FDA regulatory decisions— We are in the process of building a sales force for the planned commercialization of Neurodex. The timing and extent of these development expenditures will vary depending on the FDA’s review and decision on approval of our NDA for Neurodex. As a result, our expenses could vary significantly from quarter-to-quarter while we await regulatory decisions and complete this building process. |
| |
| | • | | Acquisitions— Our acquisition of certain rights relating to Neurodex in fiscal 2005 resulted in charges of approximately $7.2 million. We may acquire other companies or technologies, and if we do so, we will incur potentially significant charges in connection with such acquisitions and may have ongoing charges after the closing of any such transaction. Any such acquisitions could also be disruptive to our operations and may adversely affect our results of operations. |
As a result of these factors, our stock price may continue to be volatile and investors may be unable to sell their shares at a price equal to, or above, the price paid.
46
Risks Relating to Our Industry
The pharmaceutical industry is highly competitive and most of our competitors are larger and have greater resources. As a result, we face significant competitive hurdles.
The pharmaceutical and biotechnology industries are highly competitive and subject to significant and rapid technological change. We compete with hundreds of companies that develop and market products and technologies in similar areas as our research. For example, we expect that Neurodex, if approved by the FDA for marketing as a treatment of IEED/PBA, will compete against antidepressants, atypical anti-psychotic agents and other agents for the treatment of this condition.
Our competitors may have specific expertise and technologies that are better than ours and many of these companies, either alone or together with their research partners, have substantially greater financial resources, larger research and development staffs and substantially greater experience than we do. Accordingly, our competitors may successfully develop competing products. If we commence commercial sales for Neurodex™ for IEED/PBA, we may also be competing with other companies and their products with respect to manufacturing efficiencies and marketing capabilities, areas where we have limited or no direct experience.
Item 2. UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS
Not applicable.
Item 3. DEFAULTS UPON SENIOR SECURITIES
Not applicable.
Item 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
The Annual Meeting of Shareholders of AVANIR Pharmaceuticals was convened on February 6, 2006, at 9:00 a.m. local time. There were issued and outstanding on December 19, 2005, the record date, 29,012,455 shares of Class A common stock, each share being entitled to one vote, constituting all of our outstanding voting securities. There were present at the meeting in person or by proxy, our shareholders who were the holders of 25,287,673 votes of our common stock entitled to vote thereat, constituting a quorum. The following proposals received the number of votes set forth below, such votes being sufficient to pass all of the proposals.
47
Proposal No. 1: Election of Directors
| | | | | | | | | |
| Directors | | For | | | Withhold | |
| Eric K. Brandt | | | 25,145,458 | | | | 142,215 | |
| Charles A. Mathews | | | 24,609,101 | | | | 678,572 | |
| Jonathan T. Silverstein | | | 24,689,824 | | | | 597,849 | |
Proposal No. 2: Amendment to 2005 Equity Incentive Plan
| | | | | | | | | |
| | For | | Against | | Abstain | |
| | 10,367,433 | | 2,900,387 | | | | 90,734 | |
Proposal No. 4: Ratification of Selection of Deloitte & Touche LLP as Independent Registered Public Accounting Firm
| | | | | | | | | |
| | For | | Against | | Abstain | |
| | 24,841,661 | | 399,286 | | | | 46,726 | |
As we implemented a one-for-four reverse stock split prior to the Annual Meeting under the authority approved at the 2005 annual meeting, Proposal No. 3: Renewal of Authority of the Board of Directors to Effect a Reverse Stock Split was withdrawn and not voted upon at the meeting.
All of the share and vote numbers have been adjusted to reflect the one-for-four reverse stock split.
Item 5. OTHER INFORMATION
None.
Item 6. EXHIBITS
Exhibits
| | | |
| 10.1 | | Manufacturing Services Agreement between Patheon, Inc. and AVANIR Pharmaceuticals, dated January 4, 2006.* |
| | | |
| 10.2 | | Quality Agreement between Patheon, Inc. and AVANIR Pharmaceuticals, dated January 4, 2006.* |
| | | |
| 10.3 | | Docosanol License Agreement between Kobayashi Pharmaceutical Co., Ltd. and AVANIR Pharmaceuticals, dated January 5, 2006.* |
| | | |
| 15.1 | | Letter on unaudited interim financial information. |
| | | |
| 31.1 | | Certification of Principal Executive Officer Required Under Rule 13a-14(a) of the Securities Exchange Act of 1934, as amended. |
| | | |
| 31.2 | | Certification of Principal Financial Officer Required Under Rule 13a-14(a) of the Securities Exchange Act of 1934, as amended. |
| | | |
| 32.1 | | Certification of Principal Executive Officer Required Under Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. 1350. |
| | | |
| 32.2 | | Certification of Principal Financial Officer Required Under Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. 1350. |
| | |
| * | | Confidential treatment requested. |
48
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| | | | | |
| Signature | | Title | | Date |
| | | | | |
/s/ Eric K. Brandt Eric K. Brandt | | President and Chief Executive Officer (Principal Executive Officer) | | May 10, 2006 |
| | | | | |
/s/ Gregory P. Hanson, CMA Gregory P. Hanson, CMA | | Vice President, Finance and Chief Financial Officer (Principal Financial and Accounting Officer) | | May 10, 2006 |
49